
Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes


 , , and
, , and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Dataset Selection
2.2. The “Nuclear mRNA Splicing” Pathway Is Consistently Dysregulated in MS

{kind=link}
{kind=link}
{kind=link}
| Dataset (accession number) | Array Type | MS Cases/Controls | Origin | % Females (Cases/Controls) | Age (Years) | Notes on MS Patients Included | Tissue | Ref. |
|---|---|---|---|---|---|---|---|---|
| GSE21942 | Affymetrix Human Genome U133 Plus 2.0 Array | 10/15 | Finland | 100/100 | Mean age cases: 54.2 Mean age controls: 71.6 | 4 patients under treatment | Peripheral blood mononuclear cells | [20] |
| GSE41848 | Affymetrix Human Exon 1.0 ST Array | 54/38 (discovery dataset) | n.a. | 71/74 | Median age cases: 43 (range: 22–66) Median age controls: 46 (range 26–66) | Only RR-MS untreated cases included in the analyses | Whole blood | [21] |
| GSE41849 | Affymetrix Human Exon 1.0 ST Array | 21/22 (replication dataset) | 66/59 | Median age cases: 45 (range 23–61) Median age controls: 42 (range 27–61) | ||||
| GSE41890 | Affymetrix Human Gene 1.0 ST Array | 22/24 | n.a. | 56/52 | Mean age cases: 40 (range 23–66) Mean age controls: 38 (range 23–57) | RR-MS patients in remitting phase | Peripheral blood leukocytes | [22] |
| GSE17048 | Illumina HumanHT-12 V3.0 Expression BeadChip | 36/45 | Australia | 80/64 | Mean age cases: 48.5 (range 29–65) Mean age controls: 48.5 (range 23–77) | Only RR-MS untreated cases included in the analyses | Whole blood | [23,24] |
| GSE43592 | Affymetrix Human Genome U133 Plus 2.0 Array | 10/10 | Sweden | 40/40 | Mean age cases: 38 Mean age controls: 40 | RR-MS | Peripheral blood T-cells | [25] |
| GSE13732 | Affymetrix Human Genome U133 Plus 2.0 Array | 37 */28 | 92% Caucasian | 74/64 | Mean age cases: 37 Mean age controls: 35 | - | Naïve CD4+ T cells | [26] |
| Dataset (Accession Number) | No. of Identified Cluster * | No. of Significantly Enriched Clusters ** | Enrichment Score Range (max–min) *** | “Nuclear mRNA Splicing” Category | “NADH Dehydrogenase Complex” Category | ||
|---|---|---|---|---|---|---|---|
| Cluster Rank | Enrichment Score | Cluster Rank | Enrichment Score | ||||
| GSE21942 | 218 | 27 | 12–1.34 | 1 | 12 **** | 8 | 3 **** |
| GSE41848 | 246 | 37 | 4.2–1.32 | 7 | 2.67 | 6 | 2.97 |
| GSE41849 | 214 | 9 | 5.52–1.35 | - | - | 1 and 2 | 5.52 and 5.1 **** |
| GSE41890 | 185 | 10 | 5.1–1.47 | - | - | - | - |
| GSE17048 | 174 | 10 | 2.41–1.34 | 4 | 2.16 | 10 | 1.34 |
| GSE43592 | 222 | 23 | 5.24–1.32 | 10 | 1.82 | - | - |
| GSE13732 | 199 | 25 | 9.06–1.33 | 3 | 4.34 **** | - | - |
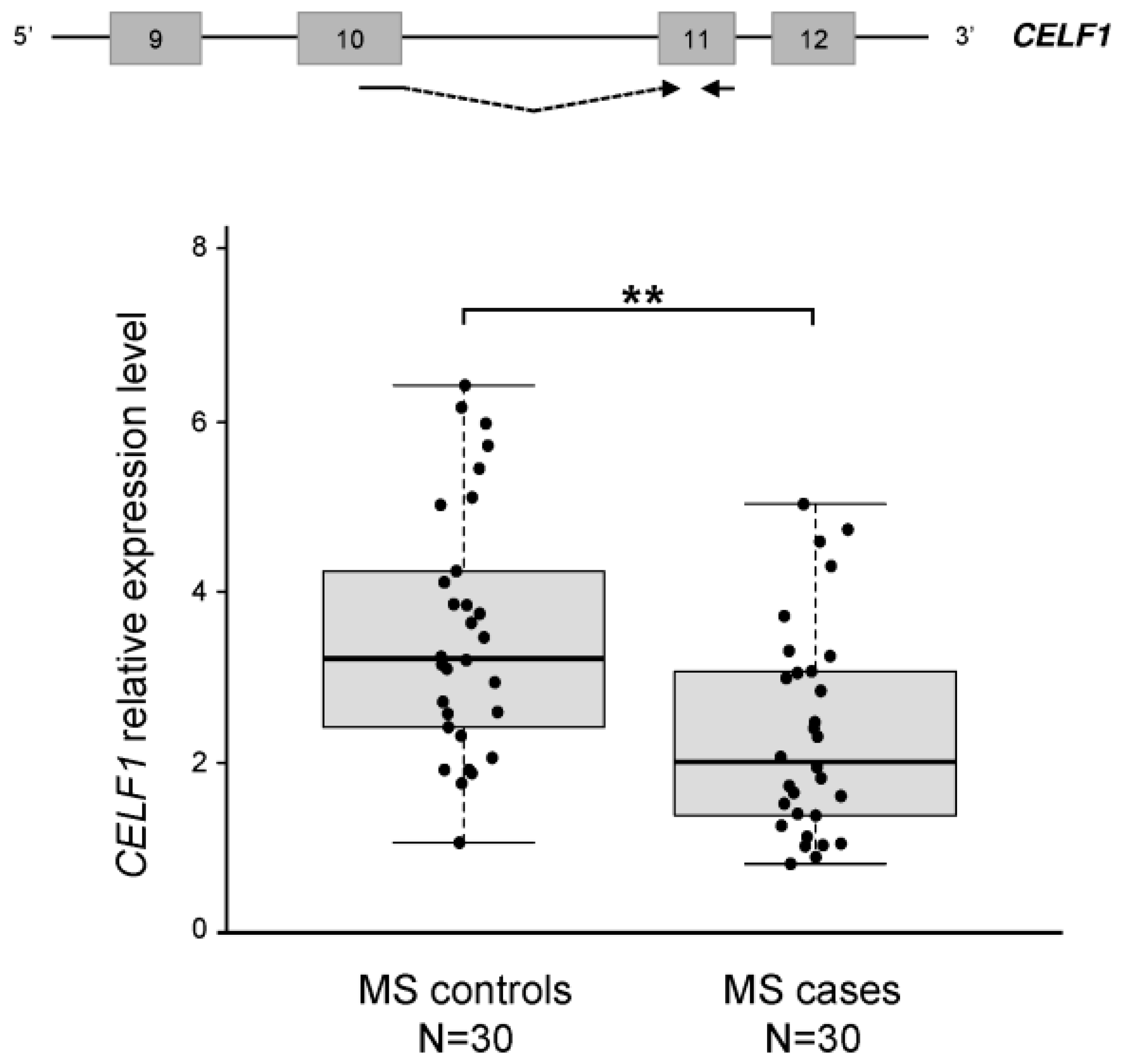
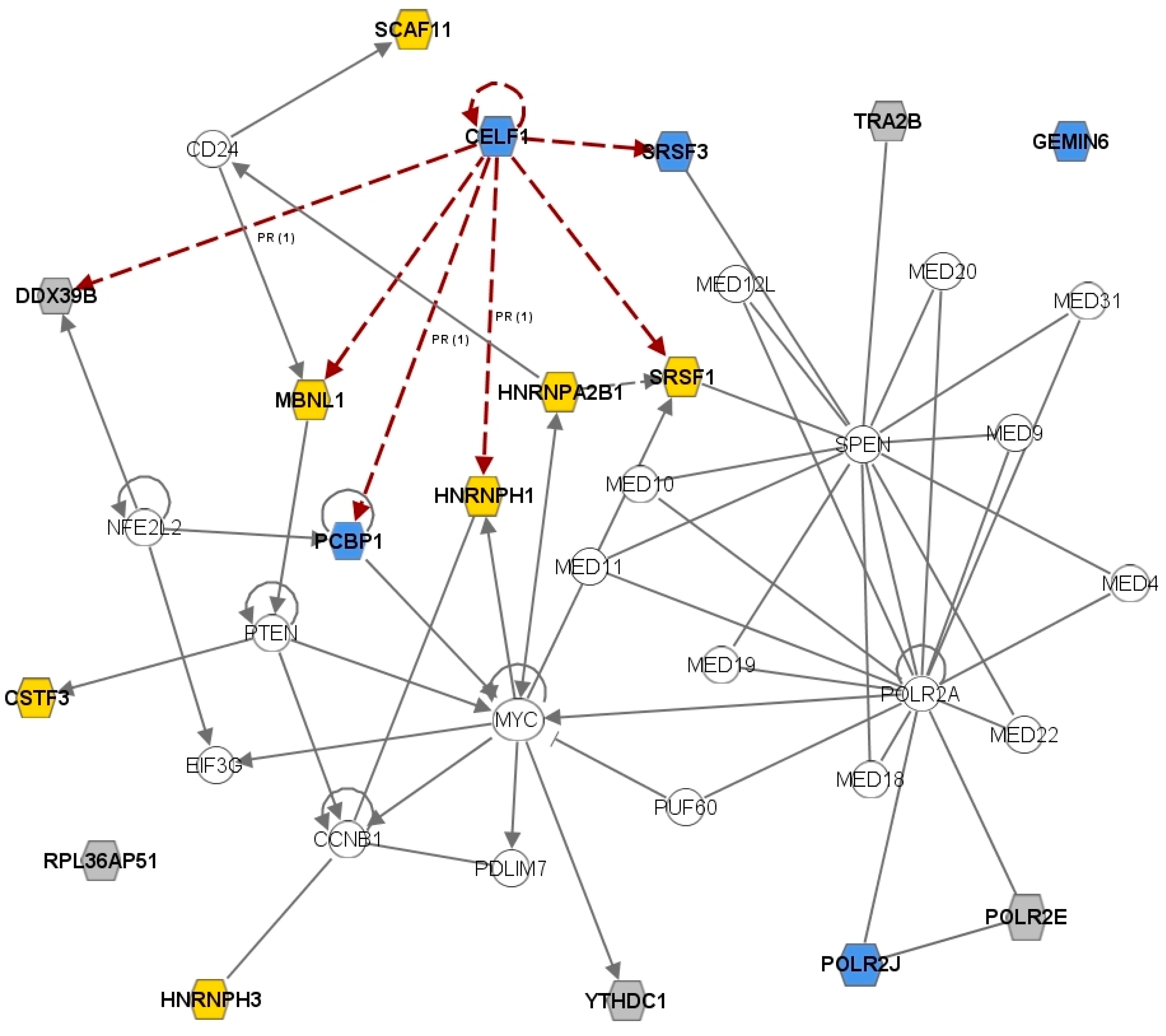
2.3. The CELF1 Gene Emerges as a Major Splicing-Regulator Altered in MS
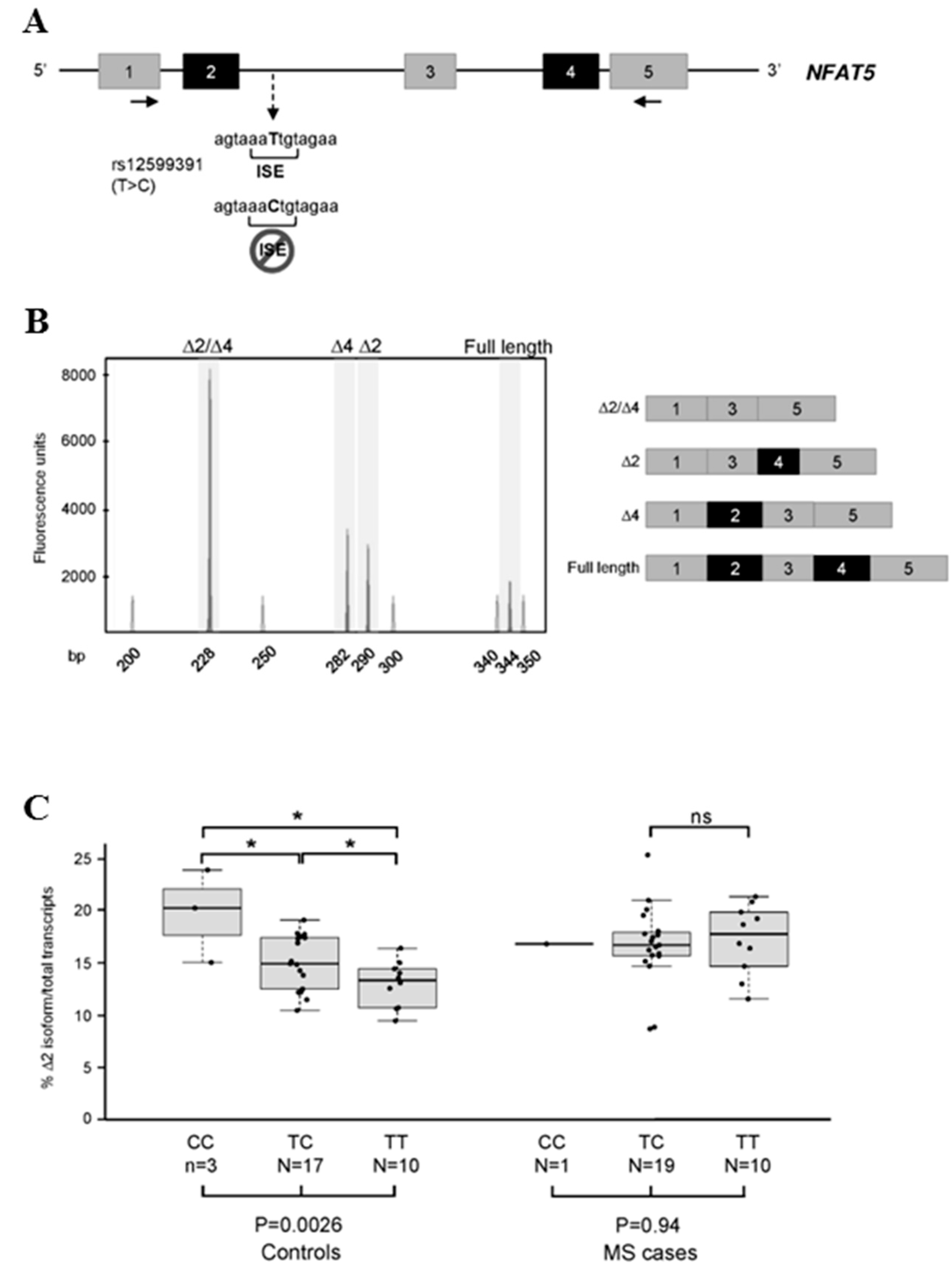
2.4. Effect of Splicing Alteration on Genotype-Dependent AS Events
| Gene Symbol | Gene Name | No. of Datasets in Which the Gene Is DE | Combined T Stat * | Combined p Value |
|---|---|---|---|---|
| CELF1/CUGBP1 | CUG triplet repeat, RNA binding protein 1 | 5 out of 5 | −38.352 | 4.0144 × 10−5 |
| MBNL1 | muscleblind-like splicing regulator 1 | 4 out of 5 | 36.567 | 7.6664 × 10−5 |
| CSTF3 | cleavage stimulation factor, 3ʹ pre-RNA, subunit 3 | 3 out of 5 | 41.24 | 1.4073 × 10−5 |
| SRSF10 | FUS interacting protein (serine/arginine-rich) 1 | n.c. | n.c. | |
| GEMIN6 | gem (nuclear organelle) associated protein 6 | −34.092 | 0.00018533 | |
| HNRNPA2B1 | heterogeneous nuclear ribonucleoprotein A2/B1 | 43.464 | 6.2483 × 10−6 | |
| HNRNPH1 | heterogeneous nuclear ribonucleoprotein H1 (H) | 39.348 | 2.7932 × 10−5 | |
| HNRNPH3 | heterogeneous nuclear ribonucleoprotein H3 (2H9) | 32.941 | 0.00027984 | |
| DDX39B | HLA-B associated transcript 1 | n.c. | n.c. | |
| PCBP1 | poly(rC) binding protein 1 | −35.428 | 0.0001146 | |
| POLR2E | polymerase (RNA) II (DNA directed) polypeptide E, 25 kDa | n.c. | n.c. | |
| POLR2J | polymerase (RNA) II (DNA directed) polypeptide J, 13.3 kDa | −27.037 | 0.0021827 | |
| RPL36AP51 | ribosomal protein L36a pseudogene 51 | n.c. | n.c. | |
| SRSF1 | splicing factor, arginine/serine-rich 1 | 46.327 | 2.1799 × 10−6 | |
| SCAF11 | splicing factor, arginine/serine-rich 2, interacting protein | 26.307 | 0.0028008 | |
| SRSF3 | splicing factor, arginine/serine-rich 3 | −42.054 | 1.0493 × 10−5 | |
| YTHDC1 | YTH domain containing 1 | n.c. | n.c. |


2.5. Discussion

3. Experimental Section
3.1. Dataset Retrieving and Analyses
3.2. MS Cases and Healthy Controls
3.3. RNA and DNA Samples
3.4. Semi-Quantitative Real-Time RT-PCR and Competitive Fluorescent RT-PCR
3.5. Genotyping of the rs12599391 Polymorphism in the NFAT5 Gene
3.6. Web Resources
4. Conclusions
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Green, R.E.; Brenner, S.E. Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc. Natl. Acad. Sci. USA 2003, 100, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J. Multiple sclerosis—A global disorder and still poorly managed. Lancet Neurol. 2008, 7, 1078–1079. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium; Hafler, D.A.; Compston, A.; Sawcer, S.; Lander, E.S.; Daly, M.J.; de Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsopoulos, N.A.; Bayer Pharma MS Genetics Working Group; Steering Committees of Studies Evaluating IFNβ-1b and a CCR1-Antagonist; ANZgene Consortium; GeneMSA; International Multiple Sclerosis Genetics Consortium; Esposito, F.; Reischl, J.; Lehr, S.; Bauer, D.; et al. Genome-wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Apperson, M.L.; Ander, B.P.; Liu, D.; Stomova, B.S.; Jickling, G.C.; Enriquez, R.; Agius, M.A.; Sharp, F.R. Differences in exon expression and alternatively spliced genes in blood of multiple sclerosis compared to healthy control subjects. J. Neuroimmunol. 2011, 230, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Evsyukova, I.; Somarelli, J.A.; Gregory, S.G.; Garcia-Blanco, M.A. Alternative splicing in multiple sclerosis and other autoimmune diseases. RNA Biol. 2010, 7, 462–473. [Google Scholar] [CrossRef] [PubMed]
- Paraboschi, E.M.; Rimoldi, V.; Soldà, G.; Tabaglio, T.; Dall’Osso, C.; Saba, E.; Vigliano, M.; Salviati, A.; Leone, M.; Benedetti, M.D.; et al. Functional variations modulating PRKCA expression and alternative splicing predispose to multiple sclerosis. Hum. Mol. Genet. 2014, 23, 6746–6761. [Google Scholar] [CrossRef] [PubMed]
- Královicová, J.; Houngninou-Molango, S.; Krämer, A.; Vorechovsky, I. Branch site haplotypes that control alternative splicing. Hum. Mol. Genet. 2004, 13, 3189–3202. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chen, I.H.; Xiong, Z.; Yan, Y.; Wang, H.; Yang, X.F. Model of stimulation-responsive splicing and strategies in identification of immunogenic isoforms of tumor antigens and autoantigens. Clin. Immunol. 2006, 121, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Hassfeld, W.; Steiner, G.; Studnicka-Benke, A.; Skriner, K.; Graninger, W.; Fischer, I.; Smolen, J.S. Autoimmune response to the spliceosome. An immunologic link between rheumatoid arthritis, mixed connective tissue disease, and systemic lupus erythematosus. Arthritis Rheumatol. 1995, 38, 777–785. [Google Scholar] [CrossRef]
- Jones, D.A.; Yawalkar, N.; Suh, K.Y.; Sadat, S.; Rich, B.; Kupper, T.S. Identification of autoantigens in psoriatic plaques using expression cloning. J. Investig. Dermatol. 2004, 123, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Yukitake, M.; Sueoka, E.; Sueoka-Aragane, N.; Sato, A.; Ohashi, H.; Yakushiji, Y.; Saito, M.; Osame, M.; Izumo, S.; Kuroda, Y. Significantly increased antibody response to heterogeneous nuclear ribonucleoproteins in cerebrospinal fluid of multiple sclerosis patients but not in patients with human T-lymphotropic virus type I-associated myelopathy/tropical spastic paraparesis. J. Neurovirol. 2008, 14, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Bekenstein, U.; Soreq, H. Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: From structural insights to post-transcriptional regulatory roles. Mol. Cell. Neurosci. 2013, 56, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Xu, L.; Shin, Y.; Gardner, L.; Hartzes, A.; Dohan, F.C.; Raine, C.; Homayouni, R.; Levin, M.C. A potential link between autoimmunity and neurodegeneration in immune-mediated neurological disease. J. Neuroimmunol. 2011, 235, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Kemppinen, A.K.; Kaprio, J.; Palotie, A.; Saarela, J. Systematic review of genome-wide expression studies in multiple sclerosis. BMJ Open 2011, 1, e000053. [Google Scholar] [CrossRef] [PubMed]
- Nickles, D.; Chen, H.P.; Li, M.M.; Khankhanian, P.; Madireddy, L.; Caillier, S.J.; Santaniello, A.; Cree, B.A.; Pelletier, D.; Hauser, S.L.; et al. Blood RNA profiling in a large cohort of multiple sclerosis patients and healthy controls. Hum. Mol. Genet. 2013, 22, 4194–4205. [Google Scholar] [CrossRef] [PubMed]
- Irizar, H.; Muñoz-Culla, M.; Sepúlveda, L.; Sáenz-Cuesta, M.; Prada, Á.; Castillo-Triviño, T.; Zamora-López, G.; de Munain, A.L.; Olascoaga, J.; Otaegui, D. Transcriptomic profile reveals gender-specific molecular mechanisms driving multiple sclerosis progression. PLoS ONE 2014, 9, e90482. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, K.S.; McKay, F.C.; Cox, M.; Riveros, C.; Armstrong, N.; Heard, R.N.; Vucic, S.; Williams, D.W.; Stankovich, J.; Brown, M.; et al. The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 2010, 19, 2134–2143. [Google Scholar] [CrossRef] [PubMed]
- Riveros, C.; Mellor, D.; Gandhi, K.S.; McKay, F.C.; Cox, M.B.; Berretta, R.; Vaezpour, S.Y.; Inostroza-Ponta, M.; Broadley, S.A.; Heard, R.N.; et al. A transcription factor map as revealed by a genome-wide gene expression analysis of whole-blood mRNA transcriptome in multiple sclerosis. PLoS ONE 2010, 5, e14176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernås, M.; Malmeström, C.; Axelsson, M.; Nookaew, I.; Wadenvik, H.; Lycke, J.; Olsson, B. MicroRNA regulate immune pathways in T-cells in multiple sclerosis (MS). BMC Immunol. 2013, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Corvol, J.C.; Pelletier, D.; Henry, R.G.; Caillier, S.J.; Wang, J.; Pappas, D.; Casazza, S.; Okuda, D.T.; Hauser, S.L.; Oksenberg, J.R.; et al. Abrogation of T cell quiescence characterizes patients at high risk for multiple sclerosis after the initial neurological event. Proc. Natl. Acad. Sci. USA 2008, 105, 11839–11844. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Gamazon, E.R.; Rebman, E.; Lee, Y.; Lee, S.; Dolan, M.E.; Cox, N.J.; Lussier, Y.A. Variants affecting exon skipping contribute to complex traits. PLoS Genet. 2012, 8, e1002998. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Cody, N.A.; Jog, S.; Biancolella, M.; Wang, T.T.; Treacy, D.J.; Luo, S.; Schroth, G.P.; Housman, D.E.; Reddy, S.; et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell 2012, 150, 710–724. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Ward, A.J.; Cherone, J.M.; Giudice, J.; Wang, T.T.; Treacy, D.J.; Lambert, N.J.; Freese, P.; Saxena, T.; Cooper, T.A.; et al. Antagonistic regulation of mRNA expression and splicing by CELF and MBNL proteins. Genome Res. 2015, 25, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Milo, R.; Miller, A. Revised diagnostic criteria of multiple sclerosis. Autoimmun. Rev. 2014, 13, 518–524. [Google Scholar] [CrossRef] [PubMed]
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Brynedal, B.; Khademi, M.; Wallström, E.; Hillert, J.; Olsson, T.; Duvefelt, K. Gene expression profiling in multiple sclerosis: A disease of the central nervous system, but with relapses triggered in the periphery? Neurobiol. Dis. 2010, 37, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Li, J.; Bumgarner, R.E. Sample size for detecting differentially expressed genes in microarray experiments. BMC Genom. 2004, 5, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Schizophrenia Consortium; Purcell, S.M.; Wray, N.R.; Stone, J.L.; Visscher, P.M.; O’Donovan, M.C.; Sullivan, P.F.; Sklar, P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.; McDonough, J.; Yin, X.; Peterson, J.; Chang, A.; Torres, T.; Gudz, T.; Macklin, W.B.; Lewis, D.A.; Fox, R.J.; et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006, 59, 478–489. [Google Scholar] [CrossRef] [PubMed]
- Van Horssen, J.; Witte, M.E.; Ciccarelli, O. The role of mitochondria in axonal degeneration and tissue repair in MS. Mult. Scler. 2012, 18, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Verkhovskaya, M.; Bloch, D.A. Energy-converting respiratory Complex I: On the way to the molecular mechanism of the proton pump. Int. J. Biochem. Cell Biol. 2013, 45, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Talla, V.; Yu, H.; Chou, T.H.; Porciatti, V.; Chiodo, V.; Boye, S.L.; Hauswirth, W.W.; Lewin, A.S.; Guy, J. NADH-dehydrogenase type-2 suppresses irreversible visual loss and neurodegeneration in the EAE animal model of MS. Mol. Ther. 2013, 21, 1876–1888. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Lee, Y.; Yamashita, T.; Yagi, T.; Iwata, S.; Cameron, A.D.; Maher, M.J. The structure of the yeast NADH dehydrogenase (Ndi1) reveals overlapping binding sites for water- and lipid-soluble substrates. Proc. Natl. Acad. Sci. USA 2012, 109, 15247–15252. [Google Scholar] [CrossRef] [PubMed]
- Poulos, M.G.; Batra, R.; Charizanis, K.; Swanson, M.S. Developments in RNA splicing and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a000778. [Google Scholar] [CrossRef] [PubMed]
- Masuda, A.; Andersen, H.S.; Doktor, T.K.; Okamoto, T.; Ito, M.; Andresen, B.S.; Ohno, K. CUGBP1 and MBNL1 preferentially bind to 3’ UTRs and facilitate mRNA decay. Sci. Rep. 2012, 2, 209. [Google Scholar] [CrossRef] [PubMed]
- Beisang, D.; Rattenbacher, B.; Vlasova-St Louis, I.A.; Bohjanen, P.R. Regulation of CUG-binding protein 1 (CUGBP1) binding to target transcripts upon T cell activation. J. Biol. Chem. 2012, 287, 950–960. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ferraris, J.D.; Klein, J.D.; Sands, J.M.; Burg, M.B.; Zhou, X. PKC-α contributes to high NaCl-induced activation of NFAT5 (TonEBP/OREBP) through MAPK ERK1/2. Am. J. Physiol. Ren. Physiol. 2015, 308, F140–F148. [Google Scholar] [CrossRef] [PubMed]
- Saarela, J.; Kallio, S.P.; Chen, D.; Montpetit, A.; Jokiaho, A.; Choi, E.; Asselta, R.; Bronnikov, D.; Lincoln, M.R.; Sadovnick, A.D.; et al. PRKCA and multiple sclerosis: Association in two independent populations. PLoS Genet. 2006, 2, e42. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Manzel, A.; Titze, J.; Kvakan, H.; Yosef, N.; Linker, R.A.; Muller, D.N.; Hafler, D.A. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 2013, 496, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Rudnev, D.; Evangelista, C.; Kim, I.F.; Soboleva, A.; Tomashevsky, M.; Marshall, K.A.; et al. NCBI GEO: Archive for high-throughput functional genomic data. Nucleic Acids Res. 2009, 37, D885–D890. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The R Project for Statistical Computing. Available online: http://www.R-project.org/ (accessed on 15 September 2015).
- Xia, J.; Fjell, C.D.; Mayer, M.L.; Pena, O.M.; Wishart, D.S.; Hancock, R.E. INMEX—A web-based tool for integrative meta-analysis of expression data. Nucleic Acids Res. 2013, 41, W63–W70. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; de Preter, K.; Pattyn, F.; Poppe, B.; van Roy, N.; de Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM, Online Mendelian Inheritance in Man. Available online: http://www.ncbi.nlm.nih.gov/omim (accessed on 15 July 2015).
- GEO DataSets, Gene Expression Omnibus repository. Available online: http://www.ncbi.nlm.nih.gov/gds/ (accessed on 24 September 2014).
- DAVID Bioinformatics Resources 6.7, The Database for Annotation, Visualization and Integrated Discovery. Available online: http://david.abcc.ncifcrf.gov/ (accessed on 20 November 2014).
- The University of California Santa Cruz Genome Browser (UCSC). Available online: http://genome.ucsc.edu/ (accessed on 15 July 2015).
- KEGG, Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed on 15 July 2015).
- INMEX, Integrative Meta-analysis of Expression Data. Available online: http://www.inmex.ca/INMEX/ (accessed on 17 March 2015).
- Markus, M.A.; Marques, F.Z.; Morris, B.J. Resveratrol, by modulating RNA processing factor levels, can influence the alternative splicing of pre-mRNAs. PLoS ONE 2011, 6, e28926. [Google Scholar] [CrossRef] [PubMed]
- Fonseca-Kelly, Z.; Nassrallah, M.; Uribe, J.; Khan, R.S.; Dine, K.; Dutt, M.; Shindler, K.S. Resveratrol neuroprotection in a chronic mouse model of multiple sclerosis. Front. Neurol. 2012, 3, 84. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Martinez, N.E.; Shahid, M.; Rose, J.W.; Carlson, N.G.; Tsunoda, I. Resveratrol exacerbates both autoimmune and viral models of multiple sclerosis. Am. J. Pathol. 2013, 183, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Li, X.; Chiriac, A.; Goel, K.; Terzic, A.; Perez-Terzic, C.; Nelson, T.J. Transcriptome from circulating cells suggests dysregulated pathways associated with long-term recurrent events following first-time myocardial infarction. J. Mol. Cell. Cardiol. 2014, 74, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Elashoff, M.R.; Wingrove, J.A.; Beineke, P.; Daniels, S.E.; Tingley, W.G.; Rosenberg, S.; Voros, S.; Kraus, W.E.; Ginsburg, G.S.; Schwartz, R.S.; et al. Development of a blood-based gene expression algorithm for assessment of obstructive coronary artery disease in non-diabetic patients. BMC Med. Genom. 2011, 4, 26. [Google Scholar] [CrossRef] [PubMed]
- Sinnaeve, P.R.; Donahue, M.P.; Grass, P.; Seo, D.; Vonderscher, J.; Chibout, S.D.; Kraus, W.E.; Sketch, M., Jr.; Nelson, C.; Ginsburg, G.S.; et al. Gene expression patterns in peripheral blood correlate with the extent of coronary artery disease. PLoS ONE 2009, 4, e7037. [Google Scholar] [CrossRef] [PubMed]
- Grayson, B.L.; Wang, L.; Aune, T.M. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun. 2011, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paraboschi, E.M.; Cardamone, G.; Rimoldi, V.; Gemmati, D.; Spreafico, M.; Duga, S.; Soldà, G.; Asselta, R. Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes. Int. J. Mol. Sci. 2015, 16, 23463-23481. https://doi.org/10.3390/ijms161023463
Paraboschi EM, Cardamone G, Rimoldi V, Gemmati D, Spreafico M, Duga S, Soldà G, Asselta R. Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes. International Journal of Molecular Sciences. 2015; 16(10):23463-23481. https://doi.org/10.3390/ijms161023463
Chicago/Turabian StyleParaboschi, Elvezia Maria, Giulia Cardamone, Valeria Rimoldi, Donato Gemmati, Marta Spreafico, Stefano Duga, Giulia Soldà, and Rosanna Asselta. 2015. "Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes" International Journal of Molecular Sciences 16, no. 10: 23463-23481. https://doi.org/10.3390/ijms161023463
APA StyleParaboschi, E. M., Cardamone, G., Rimoldi, V., Gemmati, D., Spreafico, M., Duga, S., Soldà, G., & Asselta, R. (2015). Meta-Analysis of Multiple Sclerosis Microarray Data Reveals Dysregulation in RNA Splicing Regulatory Genes. International Journal of Molecular Sciences, 16(10), 23463-23481. https://doi.org/10.3390/ijms161023463