
Cytokines and MicroRNAs as Candidate Biomarkers for Systemic Lupus Erythematosus

Abstract
:1. Introduction
2. Cytokines: Functions and Contributions to the Pathogenesis of SLE
3. MicroRNA-Functions and Contribution to the Pathogenesis of SLE

4. Cytokines as Biomarkers for SLE
4.1. IFN and IFN-Inducible Genes
4.2. IL-6
4.3. B Lymphocyte Stimulator Protein (BLys)
4.4. A Proliferation Inducing ligand (APRIL)
4.5. IL-10
4.6. IL-17 and IL-23
4.7. Tumor Necrosis Factor (TNF)
4.8. IL-12
4.9. Limitation of Cytokine Measurement
5. MicroRNA as Biomarkers for SLE
5.1. MiR-146a
5.2. MiR-125a
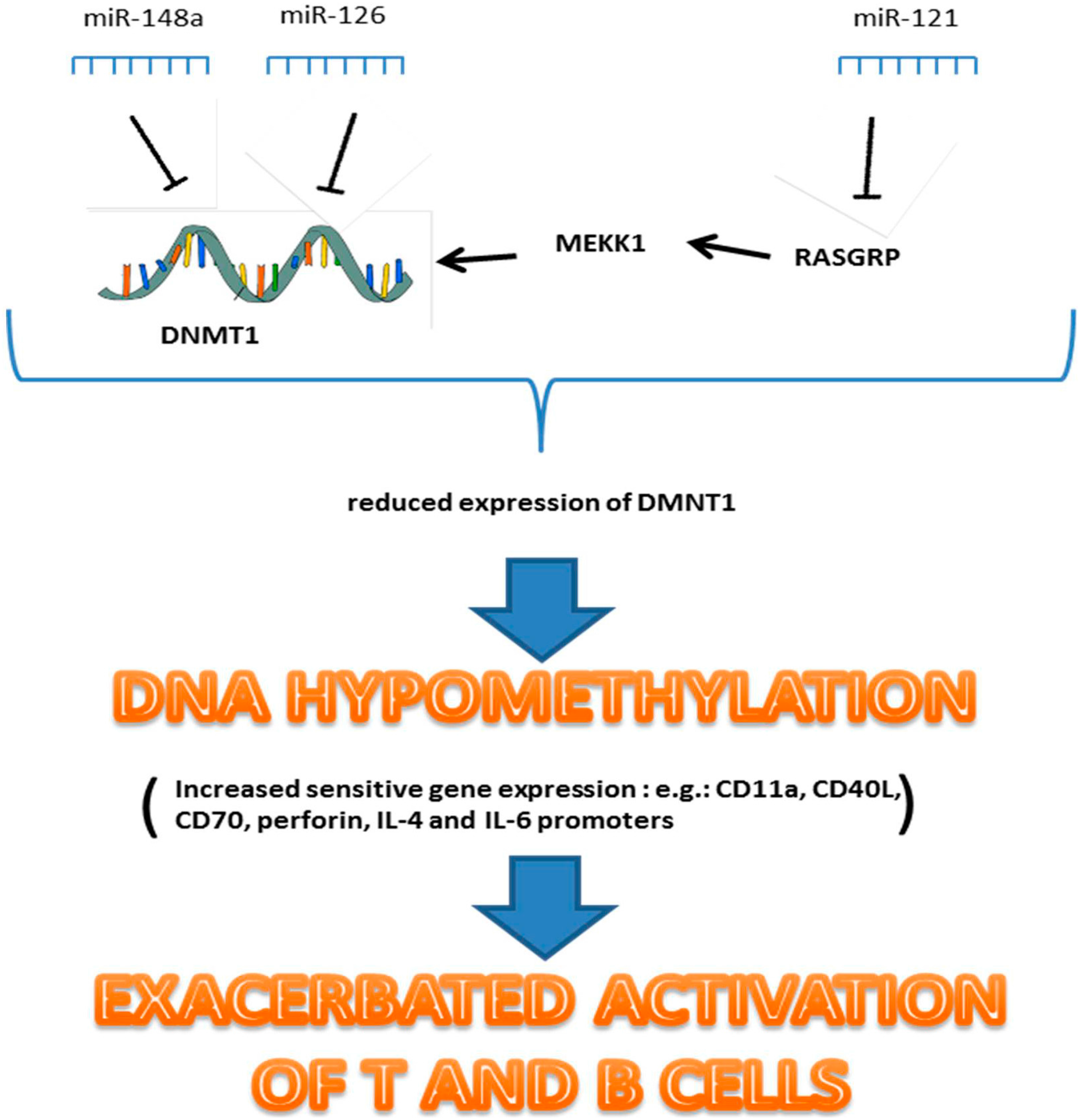
5.3. MiR-126, MiR-21 and MiR-148a
5.4. MiR-142-3p and MiR-142-5p

{kind=link}
| Biomarker | Expression in Lupus | Correlation with Lupus Activity and SLEDAI | SLE Disease Association | Ref. |
|---|---|---|---|---|
| IFN | ↑ (serum) | Positively correlated | Increase expression of auto-antigens, Central nervous system (CNS) lupus, fever | [54,55] |
| IFN inducible genes | ↑ (serum, urine) | Positively correlated with SLEDAI, correlated with flares and remission periods of SLE | More severe SLE course (CNS, hematologic and renal manifestations) | [56,57,58,59,60] |
| IL-6 | ↑ (serum, urine, BALF) | Positively correlated with SLEDAI | Lupus nephritis, increase anti-dsDNA level, CNS | [69,70,71,72] |
| BLyS | ↑ (serum, plasma) | Positively correlated with SELENA | Increase anti-dsDNA level, not associate with specific organ system involvement | [86,87,88,89] |
| IL-10 | ↑ BALF | Not significantly correlated with disease activity | Increase anti-dsDNA | [72,73,98] |
| IL-17 | ↑ (serum, kidneys) | Correlated with disease activity | Lupus nephritis | [101,102] |
| TNF | ↑ (serum, kidneys) | Correlated with disease activity? | Lupus nephritis | [106,107] |
| IL-12 | ↑ (serum, urine) | Lupus nephritis | [110,111] | |
| miR-146a | ↓ (CD4+ T cells, serum) ↑ (urine) | Inversely correlated with disease activity | Proteinuria, lupus nephritis, GFR, histological activity index | [112,113,114] |
| miR-125a | ↓ (CD4+ T cells, urine) | Inversely correlated with SLEDAI score | Lupus nephritis (GFR and creatinine ratio) | [51,115,116] |
| miR-126 | ↑ (PBMCs) | Positively correlated with disease activity | Induces DNA hypomethylation , not associate with specific organ system involvement | [37] |
| miR-21 | ↑ (PBMCs) | Positively correlated with SLEDAI score, correlated with flares and remission periods of SLE | Induces DNA hypomethylation , not associate with specific organ system involvement | [38,41,115], |
| miR-148a | ↑ (PBMCs) | Positively correlated with SLEDAI score | Induces DNA hypomethylation , not associate with specific organ system involvement | [38] |
| miR-142 | ↓ (PBMCs) ↑ (plasma, kidneys) | Not correlated with SLEDAI score | Inhibit T cell activity, not associate with specific organ system involvement | [6,43,117] |
| miR-181a | ↑ (plasma), ↓ (blood) | Positively correlated with SLEDAI score | Not associate with specific organ system involvement | [6,46] |
5.5. MiR-155
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Levy, D.M.; Kamphuis, S. Systemic lupus erythematosus in children and adolescents. Pediatr. Clin. N. Am. 2012, 59, 345–364. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.A.; Manson, J.J.; Ehrenstein, M.R.; Rahman, A. Fifty years of anti-ds DNA antibodies: Are we approaching journey’s end? Rheumatology 2007, 46, 1052–1056. [Google Scholar] [CrossRef] [PubMed]
- Egner, W. The use of laboratory tests in the diagnosis of SLE. J. Clin. Pathol. 2000, 53, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Reveille, J.D. Predictive value of autoantibodies for activity of systemic lupus erythematosus. Lupus 2004, 13, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Lourenço, E.V.; la Cava, A. Cytokines in systemic lupus erythematosus. Curr. Mol. Med. 2009, 9, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Carlsen, A.L.; Schetter, A.J.; Nielsen, C.T.; Lood, C.; Knudsen, S.; Voss, A.; Harris, C.C.; Hellmark, T.; Segelmark, M.; Jacobsen, S.; et al. Circulating microRNA expression profiles associated with systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Moore, P.A.; Belvedere, O.; Orr, A.; Pieri, K.; LaFleur, D.W.; Feng, P.; Soppet, D.; Charters, M.; Gentz, R.; Parmelee, D.; et al. BLyS: Member of the tumor necrosis factor family and B lymphocyte stimulator. Science 1999, 285, 260–263. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Schneider, P.; Rennert, P.; Browning, J. BAFF and APRIL: A tutorial on B cell survival. Annu. Rev. Immunol. 2003, 21, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browninga, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Boone, T.; Delaney, J.; Hawkins, N.; Kelley, M.; Ramakrishnan, M.; McCabe, S.; Qiu, W.R.; Kornuc, M.; Xia, X.Z.; et al. APRIL and TALL-I and receptors BCMA and TACI: System for regulating humoral immunity. Nat. Immunol. 2000, 1, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, K.; Oropallo, M.A.; Cancro, M.P.; Marshak-Rothstein, A. Role of type I interferons in the activation of autoreactive B cells. Immunol. Cell. Biol. 2012, 90, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Thoua, Y.; Ronco, P.; Verroust, P.; Tovey, M.; Morel-Maroger, L. The effect of exogenous interferon: Acceleration of autoimmune and renal diseases in (NZB/W) F1 mice. Clin. Exp. Immunol. 1980, 40, 373–382. [Google Scholar] [PubMed]
- Heremans, H.; Billiau, A.; Colombatti, A.; Hilgers, J.; de Somer, P. Interferon treatment of NZB mice: Accelerated progression of autoimmune disease. Infect. Immun. 1978, 21, 925–930. [Google Scholar] [PubMed]
- Klashman, D.J.; Martin, R.A.; Martínez-Maza, O.; Stevens, R.H. In vitro regulation of B cell differentiation by interleukin-6 and soluble CD23 in systemic lupus erythematosus B cell subpopulations and antigen-induced normal B cells. Arthritis Rheum. 1991, 34, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Garaud, S.; le Dantec, C.; Jousse-Joulin, S.; Hanrotel-Saliou, C.; Saraux, A.; Mageed, R.A.; Youinou, P.; Renaudineau, Y. IL-6 modulates CD5 expression in B cells from patients with lupus by regulating DNA methylation. J. Immunol. 2009, 182, 5623–5632. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Crispín, J.C.; Tsokos, G.C. IL-17-producing T cells in lupus nephritis. Lupus 2011, 20, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Kolls, J.K.; Lindén, A. Interleukin-17 family members and inflammation. Immunity 2004, 21, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Crispín, J.C.; Tsokos, G.C. Interleukin-17 and systemic lupus erythematosus: Current concepts. Clin. Exp. Immunol. 2009, 157, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, X.; Zou, H.; Chu, Y.; Li, M. Recovery of the immune balance between Th17 and regulatory T cells as a treatment for systemic lupus erythematosus. Rheumatology 2011, 50, 1366–1372. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, L.A.; Tsokos, G.C. The IL-2 defect in systemic lupus erythematosus disease has an expansive effect on host immunity. J. Biomed. Biotechnol. 2010, 2010, 740619. [Google Scholar] [CrossRef] [PubMed]
- Hervas-Stubbs, S.; Perez-Gracia, J.L.; Rouzaut, A.; Sanmamed, M.F.; le Bon, A.; Melero, I. Direct effects of type I interferons on cells of the immune system. Clin. Cancer Res. 2011, 17, 2619–2627. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sun, S.; Hwang, I.; Tough, D.F.; Sprent, J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 1998, 8, 591–599. [Google Scholar] [CrossRef]
- Bengtsson, A.A.; Sturfelt, G.; Gullstrand, B.; Truedsson, L. Induction of apoptosis in monocytes and lymphocytes by serum from patients with systemic lupus erythematosus—An additional mechanism to increased autoantigen load? Clin. Exp. Immunol. 2004, 135, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.J.; Lewis, E.E.; Shelden, E.A.; Somers, E.; Pavlic, R.; McCune, W.J.; Richardson, B.C. The apoptotic ligands TRAIL, TWEAK, and Fas ligand mediate monocyte death induced by autologous lupus T cells. J. Immunol. 2002, 169, 6020–6029. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, L.; Ambrosi, A.; Espinosa, A.; Ottosson, L.; Eloranta, M.L.; Zhou, W.; Elfving, A.; Greenfield, E.; Kuchroo, V.K.; Wahren-Herlenius, M. Interferon-α induces up-regulation and nuclear translocation of the Ro52 autoantigen as detected by a panel of novel Ro52-specific monoclonal antibodies. J. Clin. Immunol. 2008, 28, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Rose, L.M.; Latchman, D.S.; Isenberg, D.A. Bcl-2 and Fas, molecules which influence apoptosis. A possible role in systemic lupus erythematosus? Autoimmunity 1994, 17, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Jewell, A.P.; Worman, C.P.; Lydyard, P.M.; Yong, K.L.; Giles, F.J.; Goldstone, A.H. Interferon-α up-regulates bcl-2 expression and protects B-CLL cells from apoptosis in vitro and in vivo. Br. J. Haematol. 1994, 88, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Denny, M.F.; Thacker, S.; Mehta, H.; Somers, E.C.; Dodick, T.; Barrat, F.J.; McCune, W.J.; Kaplan, M.J. Interferon-α promotes abnormal vasculogenesis in lupus: A potential pathway for premature atherosclerosis. Blood 2007, 110, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Xie, H.; Chen, J.; Geng, L.; Chen, H.; Li, X.; Hou, Y.; Lu, L.; Shi, S.; Zeng, X.; et al. Activated NF-κB in bone marrow mesenchymal stem cells from systemic lupus erythematosus patients inhibits osteogenic differentiation through downregulating Smad signaling. Stem Cells Dev. 2013, 22, 668–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Schwarz, E.M.; O’Keefe, R.J.; Ma, L.; Boyce, B.F.; Xing, L. RANK signaling is not required for TNFα-mediated increase in CD11bhi osteoclast precursors but is essential for mature osteoclast formation in TNFα-mediated inflammatory arthritis. J. Bone Miner. Res. 2004, 19, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. Nature 2000, 408, 600–605. [Google Scholar] [PubMed]
- Ioannou, Y.; Isenberg, D.A. Current evidence for the induction of autoimmune rheumatic manifestations by cytokine therapy. Arthritis Rheum. 2000, 43, 1431–1442. [Google Scholar] [CrossRef]
- Felekkis, K.; Touvana, E.; Stefanou, C.; Deltas, C. MicroRNAs: A newly described class of encoded molecules that play a role in health and disease. Hippokratia 2010, 14, 236–240. [Google Scholar] [PubMed]
- Pan, Y.; Sawalha, A.H. Epigenetic regulation and the pathogenesis of systemic lupus erythematosus. Transl. Res. 2009, 153, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.; Scheinbart, L.; Strahler, J.; Gross, L.; Hanash, S.; Johnson, M. Evidence for impaired T cell DNA methylation in systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum. 1990, 33, 1665–1673. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, Y.; Liang, Y.; Zhao, M.; Long, H.; Ding, S.; Yin, H.; Lu, Q. MicroRNA-126 regulates DNA methylation in CD4+ T cells and contributes to systemic lupus erythematosus by targeting DNA methyltransferase 1. Arthritis Rheum. 2011, 63, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhu, S.; Yuan, M.; Cui, H.; Wang, L.; Luo, X.; Li, J.; Zhou, H.; Tang, Y.; Shen, N. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J. Immunol. 2010, 184, 6773–6781. [Google Scholar] [CrossRef] [PubMed]
- Carissimi, C.; Carucci, N.; Colombo, T.; Piconese, S.; Azzalin, G.; Cipolletta, E.; Citarella, F.; Barnaba, V.; Macino, G.; Fulci, V. miR-21 is a negative modulator of T-cell activation. Biochimie 2014, 107, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; He, L.; Zhang, R.; Liu, X.; Ren, Y.; Liu, Z.; Zhang, X.; Cheng, W.; Hua, Z.C. Regulation of T lymphocyte activation by microRNA-21. Mol. Immunol. 2014, 59, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Stagakis, E.; Bertsias, G.; Verginis, P.; Nakou, M.; Hatziapostolou, M.; Kritikos, H.; Iliopoulos, D.; Boumpas, D.T. Identification of novel microRNA signatures linked to human lupus disease activity and pathogenesis: MiR-21 regulates aberrant T cell responses through regulation of PDCD4 expression. Ann. Rheum. Dis. 2011, 70, 1496–1506. [Google Scholar] [CrossRef] [PubMed]
- Sheedy, F.J.; Palsson-McDermott, E.; Hennessy, E.J.; Martin, C.; O’Leary, J.J.; Ruan, Q.; Johnson, D.S.; Chen, Y.; O’Neill, L.A. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 2010, 11, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Liang, Y.; Zhao, M.; Liang, G.; Long, H.; Zhao, S.; Wang, Y.; Yin, H.; Zhang, P.; Zhang, Q.; et al. Decreased microRNA-142–3p/5p expression causes CD4+ T cell activation and B cell hyperstimulation in systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2953–2963. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.J.; Chau, J.; Ebert, P.J.; Sylvester, G.; Min, H.; Liu, G.; Braich, R.; Manoharan, M.; Soutschek, J.; Skare, P.; et al. MiR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 2007, 129, 147–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Lashine, Y.A.; Seoudi, A.M.; Salah, S.; Abdelaziz, A.I. Expression signature of microRNA-181-a reveals its crucial role in the pathogenesis of paediatric systemic lupus erythematosus. Clin. Exp. Rheumatol. 2011, 29, 351–357. [Google Scholar] [PubMed]
- Seddiki, N.; Brezar, V.; Ruffin, N.; Lévy, Y.; Swaminathan, S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology 2014, 142, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Leng, R.X.; Pan, H.F.; Qin, W.Z.; Chen, G.M.; Ye, D.Q. Role of microRNA-155 in autoimmunity. Cytokine Growth Factor Rev. 2011, 22, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.H.; Patterson, H.C.; Pham, D.H.; Kis-Toth, K.; Kaminski, D.A.; Tsokos, G.C. Deletion of microRNA-155 reduces autoantibody responses and alleviates lupus-like disease in the Faslpr mouse. Proc. Natl. Acad. Sci. USA 2013, 110, 20194–20199. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Liang, D.; Tang, Y.; Qu, B.; Cui, H.; Luo, X.; Huang, X.; Chen, S.; Higgs, B.W.; Jallal, B.; et al. Identification of microRNA-31 as a novel regulator contributing to impaired interleukin-2 production in T cells from patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 3715–3725. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Tang, Y.; Qu, B.; Cui, H.; Wang, S.; Wang, L.; Luo, X.; Huang, X.; Li, J.; Chen, S.; et al. MicroRNA-125a contributes to elevated inflammatory chemokine RANTES levels via targeting KLF13 in systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 3425–3435. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.D.; Lu, M.M.; Pan, H.F.; Ye, D.Q. Association of MicroRNA-146a with autoimmune diseases. Inflammation 2012, 35, 1525–1529. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xie, H.; Liu, W.; Hu, R.; Huang, B.; Tan, Y.F.; Xu, K.; Sheng, Z.F.; Zhou, H.D.; Wu, X.P.; et al. A novel microRNA targeting HDAC5 regulates osteoblast differentiation in mice and contributes to primary osteoporosis in humans. J. Clin. Investig. 2009, 119, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Becker-Merok, A.; Østli-Eilersten, G.; Lester, S.; Nossent, J. Circulating interferon-α2 levels are increased in the majority of patients with systemic lupus erythematosus and are associated with disease activity and multiple cytokine activation. Lupus 2013, 22, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, A.A.; Sturfelt, G.; Truedsson, L.; Blomberg, J.; Alm, G.; Vallin, H.; Rönnblom, L. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 2000, 9, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Baechler, E.C.; Batliwalla, F.M.; Karypis, G.; Gaffney, P.M.; Ortmann, W.A.; Espe, K.J.; Shark, K.B.; Grande, W.J.; Hughes, K.M.; Kapur, V.; et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 2003, 100, 2610–2615. [Google Scholar] [CrossRef] [PubMed]
- Biesen, R.; Demir, C.; Barkhudarova, F.; Grün, J.R.; Steinbrich-Zöllner, M.; Backhaus, M.; Häupl, T.; Rudwaleit, M.; Riemekasten, G.; Radbruch, A.; et al. Sialic acid-binding Ig-like lectin 1 expression in inflammatory and resident monocytes is a potential biomarker for monitoring disease activity and success of therapy in systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 1136–1145. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.; Grützkau, A.; Hirseland, H.; Huscher, D.; Dähnrich, C.; Dzionek, A.; Ozimkowski, T.; Schlumberger, W.; Enghard, P.; Radbruch, A.; et al. IFNα and its response proteins, IP-10 and SIGLEC-1, are biomarkers of disease activity in systemic lupus erythematosus. Ann. Rheum Dis. 2013, 72, 1639–1645. [Google Scholar] [CrossRef] [PubMed]
- Landolt-Marticorena, C.; Bonventi, G.; Lubovich, A.; Ferguson, C.; Unnithan, T.; Su, J.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Wither, J. Lack of association between the interferon-α signature and longitudinal changes in disease activity in systemic lupus erythematosus. Ann. Rheum Dis. 2009, 68, 1440–1446. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Singh, S.; Tesfasyone, H.; Dedrick, R.; Fry, K.; Lal, P.; Williams, G.; Bauer, J.; Gregersen, P.; Behrens, T.; et al. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus 2009, 18, 980–989. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Huang, J.; Liu, Y.; Xiao, L.; Wang, D.; Hua, B.; Tsao, B.P.; Sun, L. Identification of interferon-inducible genes as diagnostic biomarker for systemic lupus erythematosus. Clin. Rheumatol. 2015, 34, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Baechler, E.C.; Petri, M.; Batliwalla, F.M.; Crawford, D.; Ortmann, W.A.; Espe, K.J.; Li, W.; Patel, D.D.; Gregersen, P.K.; et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PLoS Med. 2006, 3, e491. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Chen, X.; Cui, H.; Guo, Y.; Chen, J.; Shen, N.; Bao, C. Association of elevated transcript levels of interferon-inducible chemokines with disease activity and organ damage in systemic lupus erythematosus patients. Arthritis Res. Ther. 2008, 10, R112. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Petri, M.; Batliwalla, F.M.; Koeuth, T.; Wilson, J.; Slattery, C.; Panoskaltsis-Mortari, A.; Gregersen, P.K.; Behrens, T.W.; Baechler, E.C. Interferon-regulated chemokines as biomarkers of systemic lupus erythematosus disease activity: A validation study. Arthritis Rheum. 2009, 60, 3098–3107. [Google Scholar] [CrossRef] [PubMed]
- Mathian, A.; Amoura, Z.; Adam, E.; Colaone, F.; Hoekman, M.F.; Dhellin, O.; Vandepapelière, P.; Haroche, J.; Piette, J.C.; Lebon, P.; et al. Active immunisation of human interferon α transgenic mice with a human interferon α Kinoid induces antibodies that neutralise interferon α in sera from patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Lauwerys, B.R.; Hachulla, E.; Spertini, F.; Lazaro, E.; Jorgensen, C.; Mariette, X.; Haelterman, E.; Grouard-Vogel, G.; Fanget, B.; Dhellin, O.; et al. Down-regulation of interferon signature in systemic lupus erythematosus patients by active immunization with interferon α-kinoid. Arthritis Rheum. 2013, 65, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Wallace, D.J.; Spindler, A.; Chindalore, V.; Kalunian, K.; Mysler, E.; Neuwelt, C.M.; Robbie, G.; White, W.I.; Higgs, B.W.; et al. Sifalimumab, a human anti-interferon-α monoclonal antibody, in systemic lupus erythematosus: A phase I randomized, controlled, dose-escalation study. Arthritis Rheum. 2013, 65, 1011–1021. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.M.; Jiang, J.; Abbas, A.R.; Morimoto, A.; Li, J.; Maciuca, R.; Townsend, M.; Wallace, D.J.; Kennedy, W.P.; Drappa, J. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: Results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012, 64, 3666–3676. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Huang, J.; Zhong, H.; Shen, N.; Faggioni, R.; Fung, M.; Yao, Y. Targeting interleukin-6 inflammatory autoimmune disease. Pharmacol. Ther. 2014, 141, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Dohi, K.; Hirata, E.; Kurumatani, N.; Horii, Y.; Shiiki, H.; Fukatsu, A.; Matsuda, T.; Hirano, T.; Kishimoto, T.; et al. Urinary levels of IL-6 in patients with active lupus nephritis. Clin. Nephrol. 1993, 40, 16–21. [Google Scholar] [PubMed]
- Chun, H.Y.; Chung, J.W.; Kim, H.A.; Yun, J.M.; Jeon, J.Y.; Ye, Y.M.; Kim, S.H.; Park, H.S.; Suh, C.H. Cytokine IL-6 and IL-10 as biomarkers in systemic lupus erythematosus. J. Clin. Immunol. 2007, 27, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Nielepkowicz-Goździńska, A.; Fendler, W.; Robak, E.; Kulczycka-Siennicka, L.; Górski, P.; Pietras, T.; Brzeziańska, E.; Antczak, A. Exhaled cytokines in systemic lupus erythematosus with lung involvement. Pol. Arch. Med. Wewn. 2013, 123, 141–148. [Google Scholar] [PubMed]
- Gröndal, G.; Gunnarsson, I.; Rönnelid, J.; Rogberg, S.; Klareskog, L.; Lundberg, I. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin. Exp. Rheumatol. 2000, 18, 565–570. [Google Scholar] [PubMed]
- Adhya, Z.; Borozdenkova, S.; Karim, M.Y. The role of cytokines as biomarkers in systemic lupus erythematosus and lupus nephritis. Nephrol. Dial. Transplant. 2011, 26, 3273–3280. [Google Scholar] [CrossRef] [PubMed]
- Hirohata, S.; Kanai, Y.; Mitsuo, A.; Tokano, Y.; Hashimoto, H.; NPSLE Research Subcommittee. Accuracy of cerebrospinal fluid IL-6 testing for diagnosis of lupus psychosis. A multicenter retrospective study. Clin. Rheumatol. 2009, 28, 1319–1323. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Gardber, D.E.; Bugelski, P.J.; Song, X.Y. Anti-interleukin -6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Cash, H.; Relle, M.; Menke, J.; Brochhausen, C.; Jones, S.A.; Topley, N.; Galle, P.R.; Schwarting, A. Interleukin 6 (IL-6) deficiency delays lupus nephritis in MRL-Faslpr mice: The IL-6 pathway as new therapeutic target in treatment of autoimmune kidney disease in systemic lupus erythematosus. J. Rheumatol. 2010, 37, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Yarboro, C.; Fischer, R.; Pham, T.H.; Lipsky, P.; Illei, G.G. Impact of anti-interleukin-6 receptor blockade on circulation T and B cells subsets in patients with systemic lupus erythematosus. Ann. Rheum. Dis. 2012, 72, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Wagge, A.; Slupphaug, G.; Shalaby, R. Glucorticosteroids inhibit the production of IL-6 from monocytes, endothelial cells and fibroblasts. Eur. J. Immunol. 1990, 20, 2439–2443. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, I.; Tanaka, H.; Nakao, M.; Shichijo, S.; Itoh, K. Nonsteroidal anti-inflammatory drugs differently regulate cytokine production in human lymphocytes: Up-regulation of TNF, INF-γ and IL-2, in contranst to down-regulation of IL-6 production. Cytokine 1995, 7, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Blanco, R.; Brzosko, M.; Burgos-Vargas, R.; Halland, A.M.; Vernon, E.; Ambs, P.; Fleischmann, R. Tocilizumab inhibits structural joint damage in rheumatoid arthritis patients with inadequate response to methotrexate: Results from the double-blind treatment phase of randomized placebo-controlled trial of tocilizumab safety and prevention of structural joint damage at one year. Arthritis Rheum. 2011, 63, 609–621. [Google Scholar] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Anti-interleukin-6 receptor antibody, tozilizumab, for the treatment of autoimmune diseses. FEBS Lett. 2011, 585, 3699–3709. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto, N.; Terao, K.; Mima, T.; Nakahara, H.; Takagi, N.; Kakehi, T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of anti-IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008, 112, 3959–3964. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Janikasashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepietowski, J.C.; Nilganuwong, S.; Wozniacka, A.; Kuhn, A.; Nyberg, F.; van Vollenhoven, R.F.; Bengtsson, A.A.; Reich, A.; de Vries, D.E.; van Hartingsveldt, B.; et al. Phase I, randomized, double-blind, placebo-controlled, multiple intravenous, dose-ascending study of sirukumab in cutaneous or systemic lupus erythematosus. Arthritis Rheum. 2013, 65, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Petri, M.; Stohl, W.; Chatham, W.; McCune, W.J.; Chevrier, M.; Ryel, J.; Recta, V.; Zhong, J.; Freimuth, W. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Metyas, S.; Tan, S.M.; Cheema, G.S.; Oamar, B.; Xu, D.; Roschke, V.; Wu, Y.; Baker, K.P.; Hilbert, D.M. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: Longitudinal observations. Arthritis Rheum. 2003, 48, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Neusser, M.A.; Lindenmeyer, M.T.; Edenhofer, I.; Gaiser, S.; Kretzler, M.; Regele, H.; Segerer, S.; Cohen, C.D. Intrarenal production of B-cell survival factors in human lupus nephritis. Mod. Pathol. 2011, 24, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, F.B.; Northcott, M.; Hoi, A.; Mackay, F.; Morand, E.F. Association of serum B cell activating factor from the tumour necrosis factor family (BAFF) and a proliferation-inducing ligand (APRIL) with central nervous system and renal disease in systemic lupus erythematosus. Lupus 2013, 22, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Zickert, A.; Sundelin, B.; Axelsson, M.; Gerhardsson, J.; Svenungsson, E.; Malmström, V.; Gunnarsson, I. Evaluation of B lymphocyte stimulator and a proliferation inducing ligand as candidate biomarkers in lupus nephritis based on clinical and histopathological outcome following induction therapy. Lupus Sci. Med. 2015, 22, e000061. [Google Scholar] [CrossRef] [PubMed]
- Manzi, S.; Sánchez-Guerrero, J.; Merrill, J.T.; Furie, R.; Gladman, D.; Navarra, S.V.; Ginzler, E.M.; D’Cruz, D.P.; Doria, A.; Cooper, S.; et al. Effects of belimumab, a B lymphocyte stimulator-specific inhibitor, on disease activity across multiple organ domains in patients with systemic lupus erythematosus: Combined results from two phase III trials. Ann. Rheum. Dis. 2012, 71, 1833–1838. [Google Scholar] [CrossRef] [PubMed]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzová, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Parodis, I.; Axelsson, M.; Gunnarsson, I. Belimumab for systemic lupus erythematosus: A practice-based view. Lupus 2013, 22, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Hopia, L.; Thangarajh, M.; Khademi, M.; Laveskog, A.; Wallström, E.; Svenungsson, E.; Andersson, M. Cerebrospinal fluid levels of a proliferation-inducing ligand (APRIL) are increased in patients with neuropsychiatric systemic lupus erythematosus. Scand. J. Rheumatol. 2011, 40, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Treamtrakanpon, W.; Tantivitayakul, P.; Benjachat, T.; Somparn, P.; Kittikowit, W.; Eiamong, S.; Leelahavanichkul, A.; Hirankarn, N.; Avihingsanon, Y. APRIL, a proliferation-inducing ligand, as a potential marker of lupus nephritis. Arthritis Res. Ther. 2012, 14, R252. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, D.A.; Gray, J.D.; Behrendsen, S.C.; Kubin, M.; Rengaraju, M.; Ohtsuka, K.; Trinchieri, G. Decreased production of interleukin-12 and other Th1-type cytokines in patients with recent-onset systemic lupus erythematosus. Arthritis Rheum. 1998, 41, 838–844. [Google Scholar] [CrossRef]
- Capper, E.R.; Maskill, J.K.; Gordon, C.; Blakemore, A.I. Interleukin (IL)-10, IL-1ra and IL-12 profiles in active and quiescent systemic lupus erythematosus: Could longitudinal studies reveal patient subgroups of differing pathology? Clin. Exp. Immunol. 2004, 138, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Houssiau, F.A.; Lefebvre, C.; Vanden Berghe, M.; Lambert, M.; Devogelaer, J.P.; Renauld, J.C. Serum interleukin 10 titers in systemic lupus erythematosus reflect disease activity. Lupus 1995, 4, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, R.; Wang, H.; Jiang, P.; Zhang, J.; Zhang, M.; Gu, L.; Yang, X.; Zhang, M.; Ji, X. Serum IL-10 from systemic lupus erythematosus patients suppresses the differentiation and function of monocyte-derived dendritic cells. J. Biomed. Res. 2012, 26, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.K.; Lit, L.C.; Tam, L.S.; Li, E.K.; Wong, P.T.; Lam, C.W. Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: Implications for Th17-mediated inflammation in auto-immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.; Lee, W.W.; Lee, S.H.; Kim, S.H.; Kang, S.W.; Craft, J.; Kang, I. Dysregulated balance of Th17 and Th1 cells in systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, R53. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Zickert, A.; Amoudruz, P.; Sundström, Y.; Rönnelid, J.; Malmström, V.; Gunnarsson, I. IL-17 and IL-23 in lupus nephritis - association to histopathology and response to treatment. BMC Immunol. 2015, 16, 7. [Google Scholar] [CrossRef] [PubMed]
- Aringer, M.; Smolen, J.S. The role of tumor necrosis factor-α in systemic lupus erythematosus. Arthritis Res. Ther. 2008, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, Y.; Kishimoto, S. Tumour necrosis factor/cachectin plays a key role in autoimmune pulmonary inflammation in lupus-prone mice. Clin. Exp. Immunol. 1991, 85, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.C.; Yui, M.A.; Wuthrich, R.P.; Kelley, V.E. Tumor necrosis factor and IL-1 in New Zealand Black/White mice. Enhanced gene expression and acceleration of renal injury. J. Immunol. 1989, 143, 3470–3475. [Google Scholar] [PubMed]
- Kontoyiannis, D.; Kollias, G. Accelerated autoimmunity and lupus nephritis in NZB mice with an engineered heterozygous deficiency in tumor necrosis factor. Eur. J. Immunol. 2000, 30, 2038–2047. [Google Scholar] [CrossRef]
- Trinchieri, G. Proinflammatory and immunoregulatory functions of interleukin-12. Int. Rev. Immunol. 1998, 16, 365–396. [Google Scholar] [CrossRef] [PubMed]
- Tokano, Y.; Morimoto, S.; Kaneko, H.; Amano, H.; Nozawa, K.; Takasaki, Y.; Hashimoto, H. Levels of IL-12 in the sera of patients with systemic lupus erythematosus (SLE)—Relation to Th1- and Th2-derived cytokines. Clin. Exp. Immunol. 1999, 116, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, C.C.; Jeschke, M.G.; Herndon, D.N.; Gamelli, R.; Gibran, N.; Klein, M.; Silver, G.; Arnoldo, B.; Remick, D.; Tompkins, R.G. Temporal cytokine profiles in severely burned patients: A comparison of adults and children. Mol. Med. 2008, 14, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, X.; Cui, H.; Ni, X.; Yuan, M.; Guo, Y.; Huang, X.; Zhou, H.; de Vries, N.; Tak, P.P.; et al. MicroRNA-146A contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum. 2009, 60, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Yang, W.; Ye, D.Q.; Cui, H.; Zhang, Y.; Hirankarn, N.; Qian, X.; Tang, Y.; Lau, Y.L.; de Vries, N.; et al. A functional variant in microRNA-146a promoter modulates its expression and confers disease risk for systemic lupus erythematosus. PLoS Genet. 2011, 7, e1002128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Te, J.L.; Dozmorov, I.M.; Guthridge, J.M.; Nguyen, K.L.; Cavett, J.W.; Kelly, J.A.; Bruner, G.R.; Harley, J.B.; Ojwang, J.O. Identification of unique microRNA signature associated with lupus nephritis. PLoS ONE 2010, 5, e10344. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, W.; Ouyang, X.; Li, W.; Dai, Y. Circulating microRNAs as candidate biomarkers in patients with systemic lupus erythematosus. Transl. Res. 2012, 160, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Abulaban, K.; Fall, N.; Nelson, S.; Witte, D.; Devarajan, P.; Bennett, M.; Brunner, H.I. MicroRNA’s role as biomarkers of lupus nephritis in children. Pediatr. Rheumatol. 2014, 12, P107. [Google Scholar] [CrossRef]
- Dai, Y.; Sui, W.; Lan, H.; Yan, Q.; Huang, H.; Huang, Y. Comprehensive analysis of microRNA expression patterns in renal biopsies of lupus nephritis patients. Rheumatol. Int. 2009, 29, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Divekar, A.A.; Dubey, S.; Gangalum, P.R.; Singh, R.R. Dicer insufficiency and microRNA-155 overexpression in lupus regulatory T cells: An apparent paradox in the setting of an inflammatory milieu. J. Immunol. 2011, 186, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Tam, L.S.; Li, E.K.; Kwan, B.C.; Chow, K.M.; Luk, C.C.; Li, P.K.; Szeto, C.C. Serum and urinary cell-free MiR-146a and MiR-155 in patients with systemic lupus erythematosus. J. Rheumatol. 2010, 37, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stypińska, B.; Paradowska-Gorycka, A. Cytokines and MicroRNAs as Candidate Biomarkers for Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2015, 16, 24194-24218. https://doi.org/10.3390/ijms161024194
Stypińska B, Paradowska-Gorycka A. Cytokines and MicroRNAs as Candidate Biomarkers for Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2015; 16(10):24194-24218. https://doi.org/10.3390/ijms161024194
Chicago/Turabian StyleStypińska, Barbara, and Agnieszka Paradowska-Gorycka. 2015. "Cytokines and MicroRNAs as Candidate Biomarkers for Systemic Lupus Erythematosus" International Journal of Molecular Sciences 16, no. 10: 24194-24218. https://doi.org/10.3390/ijms161024194
APA StyleStypińska, B., & Paradowska-Gorycka, A. (2015). Cytokines and MicroRNAs as Candidate Biomarkers for Systemic Lupus Erythematosus. International Journal of Molecular Sciences, 16(10), 24194-24218. https://doi.org/10.3390/ijms161024194