
Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis

Abstract
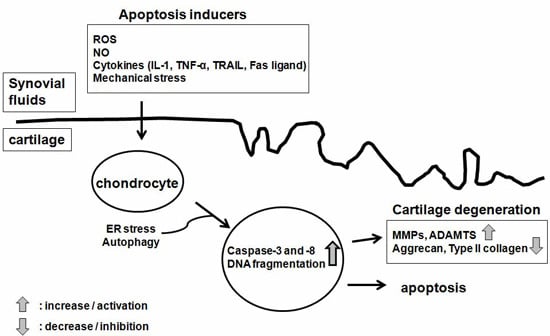
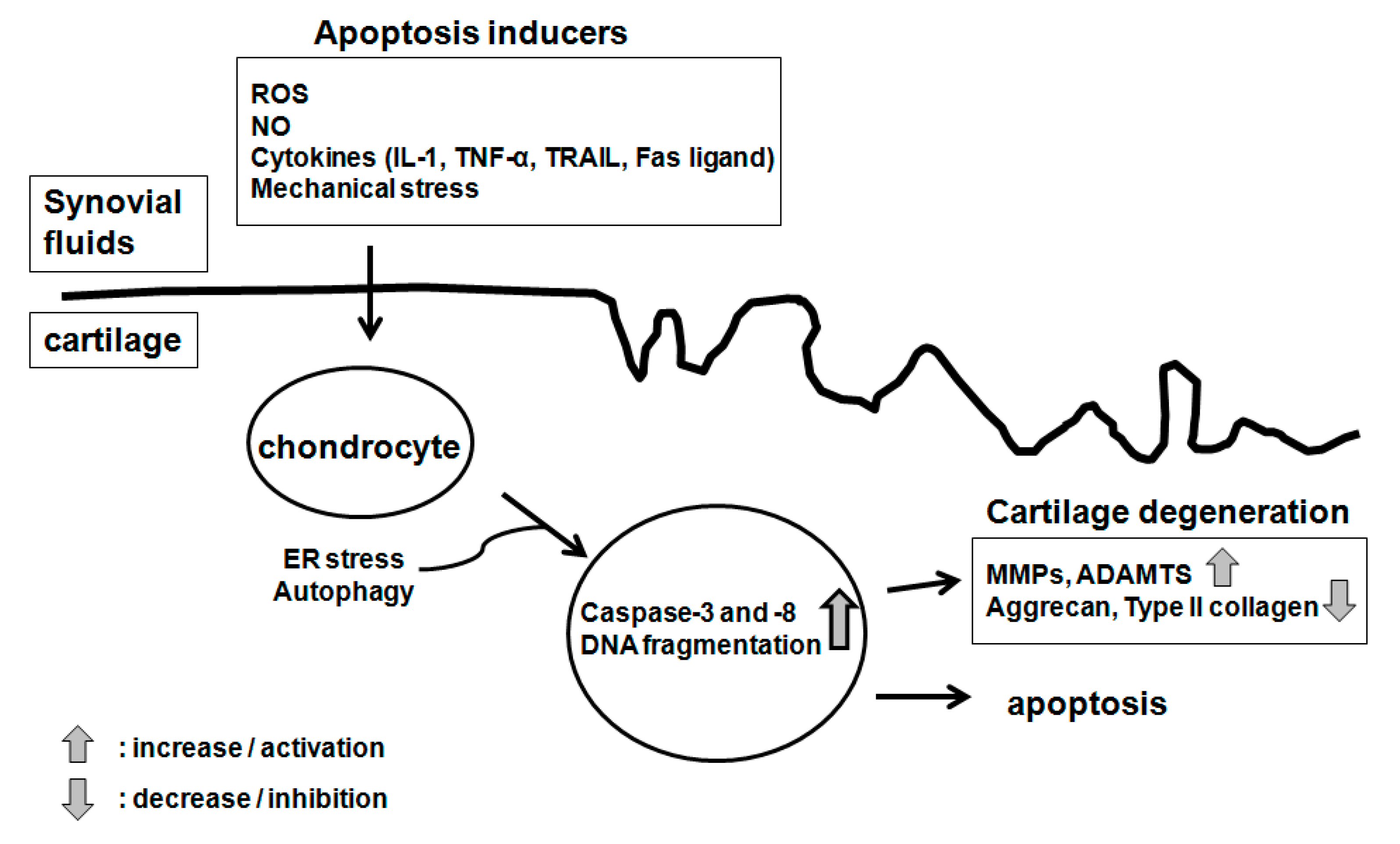
:
1. Physiological and Pathological Role of Apoptosis
2. Mediators of Apoptosis
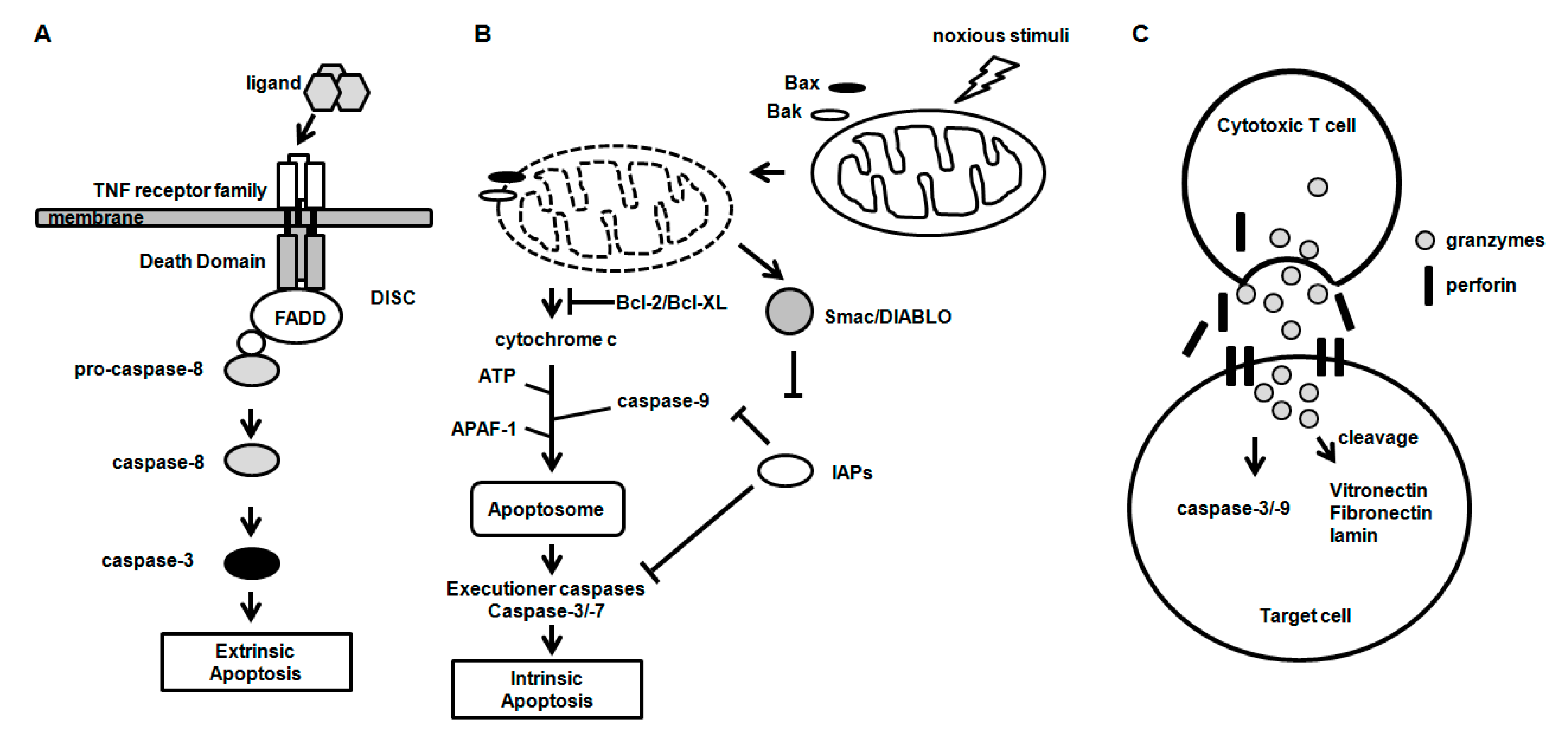
2.1. Caspases

{kind=link}
{kind=link}
{kind=link}
| Protein Name | Description |
|---|---|
| 1. Caspases family | |
| Initiator caspases | - Activated by dimerization via DED or CARD in adaptor proteins |
| Caspase-2 | |
| Caspase-8 | |
| Caspase-9 | |
| Caspase-10 | |
| Executioner caspases | - Activated by proteolysis |
| Caspase-3 | |
| Caspase-6 | |
| Caspase-7 | |
| Inflammatory caspases | - Activated by dimerization |
| Caspase-1 | |
| Caspase-4 | |
| Caspase-5 | |
| 2. Caspase inhibitors | |
| Inhibitors of apoptosis (IAP) | - Containing one or three tandem BIR domains, RING-zinc finger domain, or CARD domain |
| X-linked inhibitor of apoptosis | |
| Cellular IAP1/Human IAP2 | |
| Cellular IAP2/Human IAP1 | |
| Testis-specific IAP | |
| BIR-containing ubiquitin conjugating enzyme | |
| Survivin | |
| Livin | |
| cellular FLICE- inhibitory protein (c-FLIP) | - Containing two DED - Recruited to the DISC through DED–DED interactions |
| c-FLIPL (c-FLIP long) | |
| c-FLIPS (c-FLIP short) | |
| 3. Bcl-2 family | |
| Anti-apoptotic members | - Containing BH1, BH2, BH3, BH4, and TM domains |
| Bcl-2 | |
| Bcl-XL | |
| Bcl-W | |
| Mcl-1 | |
| A1 | |
| Multi-domain pro-apoptotic members | - Containing BH1, BH2, BH3, and TM domains |
| Bax | |
| Bak | |
| BH3-only pro-apoptotic members | - Containing BH3 domain or BH3 and TM domains |
| Bid | |
| Bad | |
| Bim | |
| Bik | |
2.2. B-Cell Chronic Lymphocytic Leukemia (CLL)/Lymphoma 2 (Bcl-2) Family Proteins
3. Apoptosis Signaling Pathways

4. The Pathogenesis of Osteoarthritis (OA)
5. Implication of Chondrocyte Apoptosis in OA Pathogenesis
6. The Mechanism of Chondrocyte Apoptosis

6.1. Molecular Inducers of Chondrocyte Apoptosis
6.2. Modulation of Mitochondrial Activity
6.3. MicroRNA and Chondrocyte Apoptosis
6.4. Chondrocyte Senescence and Apoptosis
6.5. Autophagy, Endoplasmic Reticulum (ER) Stress and Chondrocyte Apoptosis
6.6. Other Forms of Programmed Cell Death in Chondrocytes
7. Targeting Apoptosis in OA Treatment
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Hacker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, D.G. The role of apoptosis in wound healing. Int. J. Biochem. Cell Biol. 1998, 30, 1019–1030. [Google Scholar] [CrossRef]
- Tiwari, M.; Prasad, S.; Tripathi, A.; Pandey, A.N.; Ali, I.; Singh, A.K.; Shrivastav, T.G.; Chaube, S.K. Apoptosis in mammalian oocytes: A review. Apoptosis 2015, 20, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Lund, L.R.; Romer, J.; Thomasset, N.; Solberg, H.; Pyke, C.; Bissell, M.J.; Dano, K.; Werb, Z. Two distinct phases of apoptosis in mammary gland involution: Proteinase-independent and -dependent pathways. Development 1996, 122, 181–193. [Google Scholar] [PubMed]
- Croce, C.M. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial gateways to cancer. Mol. Asp. Med. 2010, 31, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Delbridge, A.R.; Strasser, A. The Bcl-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prev. 2015, 16, 2129–2144. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Friedman, D.J.; Wang, C.; Metelev, V.; Pardee, A.B. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science 1995, 268, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Hroudova, J.; Singh, N.; Fisar, Z. Mitochondrial dysfunctions in neurodegenerative diseases: Relevance to Alzheimer’s disease. Biomed Res. Int. 2014, 2014, 175062. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, V.; Wu, Y.; Boonplueang, R.; Chen, S.; Stevenson, F.F.; Yantiri, F.; Yang, L.; Beal, M.F.; Andersen, J.K. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s disease. J. Neurosci. 2001, 21, 9519–9528. [Google Scholar] [PubMed]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [PubMed]
- Pop, C.; Salvesen, G.S. Human caspases: Activation, specificity, and regulation. J. Biol. Chem. 2009, 284, 21777–21781. [Google Scholar] [CrossRef] [PubMed]
- Howley, B.; Fearnhead, H.O. Caspases as therapeutic targets. J. Cell. Mol. Med. 2008, 12, 1502–1516. [Google Scholar] [CrossRef] [PubMed]
- Labbe, K.; Saleh, M. Cell death in the host response to infection. Cell Death Differ. 2008, 15, 1339–1349. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Hunter, A.M.; LaCasse, E.C.; Korneluk, R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis 2007, 12, 1543–1568. [Google Scholar] [CrossRef] [PubMed]
- Eckelman, B.P.; Salvesen, G.S. The human anti-apoptotic proteins cIAP1 and cIAP2 bind but do not inhibit caspases. J. Biol. Chem. 2006, 281, 3254–3260. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, J.W.; Varfolomeev, E.; Goncharov, T.; Fedorova, A.V.; Kirkpatrick, D.S.; Izrael-Tomasevic, A.; Phu, L.; Arnott, D.; Aghajan, M.; Zobel, K.; et al. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2(1). Biochem. J. 2009, 417, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grutter, M.G. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef] [PubMed]
- Van Delft, M.F.; Huang, D.C. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006, 16, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.M.; Cory, S. Life-or-death decisions by the Bcl-2 protein family. Trends Biochem. Sci. 2001, 26, 61–66. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak peptide complex: Recognition between regulators of apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Dai, S.; Zhu, Y.; Marrack, P.; Kappler, J.W. The structure of a Bcl-xL/Bim fragment complex: Implications for Bim function. Immunity 2003, 19, 341–352. [Google Scholar] [CrossRef]
- Hinds, M.G.; Lackmann, M.; Skea, G.L.; Harrison, P.J.; Huang, D.C.; Day, C.L. The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J. 2003, 22, 1497–1507. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M. The apoptosome: Heart and soul of the cell death machine. Neoplasia 1999, 1, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; van Gurp, M.; Depuydt, B.; Srinivasula, S.M.; Rodriguez, I.; Alnemri, E.S.; Gevaert, K.; Vandekerckhove, J.; Declercq, W.; Vandenabeele, P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002, 9, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Schimmer, A.D. Inhibitor of apoptosis proteins: Translating basic knowledge into clinical practice. Cancer Res. 2004, 64, 7183–7190. [Google Scholar] [CrossRef] [PubMed]
- Subasinghe, W.; Syed, I.; Kowluru, A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: Evidence for regulation by Rac1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R12–R20. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, P.R.; Granville, D.J. Granzyme B in injury, inflammation, and repair. Trends Mol. Med. 2012, 18, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Buzza, M.S.; Zamurs, L.; Sun, J.; Bird, C.H.; Smith, A.I.; Trapani, J.A.; Froelich, C.J.; Nice, E.C.; Bird, P.I. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J. Biol. Chem. 2005, 280, 23549–23558. [Google Scholar] [CrossRef] [PubMed]
- Park, N.R.; Lim, K.E.; Han, M.S.; Che, X.; Park, C.; Kim, J.E.; Taniuchi, I.; Bae, S.C.; Choi, J.Y. Core binding factor β plays a critical role during chondrocyte differentiation. J. Cell. Physiol. 2016, 231, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Rabie, A.B.; Tang, G.H.; Hagg, U. Cbfa1 couples chondrocytes maturation and endochondral ossification in rat mandibular condylar cartilage. Arch. Oral Biol. 2004, 49, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hoemann, C.D.; Tran-Khanh, N.; Chevrier, A.; Chen, G.; Lascau-Coman, V.; Mathieu, C.; Changoor, A.; Yaroshinsky, A.; McCormack, R.G.; Stanish, W.D.; et al. Chondroinduction is the main cartilage repair response to microfracture and microfracture with BST-CarGel: Results as shown by ICRS-II histological scoring and a novel zonal collagen type scoring method of human clinical biopsy specimens. Am. J. Sports Med. 2015. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Tran, K.; Zhou, G.; Bedi, A.; Shelke, N.B.; Yu, X.; Kumbar, S.G. Guided differentiation of bone marrow stromal cells on co-cultured cartilage and bone scaffolds. Soft Matter 2015, 11, 7648–7655. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, X.; Tang, S.; Zhang, X.; Feng, Z.; Cui, S. Repair of massively defected hemi-joints using demineralized osteoarticular allografts with protected cartilage. J. Mater. Sci. Mater. Med. 2015, 26, 277. [Google Scholar] [CrossRef] [PubMed]
- McMahon, L.A.; O’Brien, F.J.; Prendergast, P.J. Biomechanics and mechanobiology in osteochondral tissues. Regen. Med. 2008, 3, 743–759. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Teguh, D.; Wu, J.P.; He, B.; Kirk, T.B.; Qin, S.; Li, S.; Chen, H.; Xue, W.; Ng, B.; et al. Protein kinase C delta null mice exhibit structural alterations in articular surface, intra-articular and subchondral compartments. Arthritis Res. Ther. 2015, 17, 210. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.S.; Horton, W.E., Jr. Chondrocyte apoptosis increases with age in the articular cartilage of adult animals. Anat. Rec. 1998, 250, 418–425. [Google Scholar] [CrossRef]
- Kim, H.A.; Blanco, F.J. Cell death and apoptosis in osteoarthritic cartilage. Curr. Drug Targets 2007, 8, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Kop’eva, T.N.; Bel’skaia, O.B.; Astapenko, M.G.; Arutiunov, A.G. Morphology of articular cartilage in osteoarthrosis. Arkh. Patol. 1986, 48, 40–46. [Google Scholar] [PubMed]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: A study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheumatol. 2001, 44, 1304–1312. [Google Scholar] [CrossRef]
- Heraud, F.; Heraud, A.; Harmand, M.F. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Kim, H.A.; Roach, H.I. Apoptosis in osteoarthritis. Rheum. Dis. Clin. N. Am 2004, 30, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Jiao, K.; Zhang, J.; Zhang, M.; Wei, Y.; Wu, Y.; Qiu, Z.Y.; He, J.; Cao, Y.; Hu, J.; Zhu, H.; et al. The identification of CD163 expressing phagocytic chondrocytes in joint cartilage and its novel scavenger role in cartilage degradation. PLoS ONE 2013, 8, e53312. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Suh, D.I.; Song, Y.W. Relationship between chondrocyte apoptosis and matrix depletion in human articular cartilage. J. Rheumatol. 2001, 28, 2038–2045. [Google Scholar] [PubMed]
- Pulai, J.I.; del Carlo, M., Jr.; Loeser, R.F. The α5β1 integrin provides matrix survival signals for normal and osteoarthritic human articular chondrocytes in vitro. Arthritis Rheumatol. 2002, 46, 1528–1535. [Google Scholar] [CrossRef] [PubMed]
- Zemmyo, M.; Meharra, E.J.; Kuhn, K.; Creighton-Achermann, L.; Lotz, M. Accelerated, aging-dependent development of osteoarthritis in α1 integrin-deficient mice. Arthritis Rheumatol. 2003, 48, 2873–2880. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Ochs, R.L.; Schwarz, H.; Lotz, M. Chondrocyte apoptosis induced by nitric oxide. Am. J. Pathol. 1995, 146, 75–85. [Google Scholar] [PubMed]
- Borge, L.; Demignot, S.; Adolphe, M. Type II transglutaminase expression in rabbit articular chondrocytes in culture: Relation with cell differentiation, cell growth, cell adhesion and cell apoptosis. Biochim. Biophys. Acta 1996, 1312, 117–124. [Google Scholar] [CrossRef]
- Hashimoto, S.; Setareh, M.; Ochs, R.L.; Lotz, M. Fas/Fas ligand expression and induction of apoptosis in chondrocytes. Arthritis Rheumatol. 1997, 40, 1749–1755. [Google Scholar] [CrossRef]
- Ishizaki, Y.; Burne, J.F.; Raff, M.C. Autocrine signals enable chondrocytes to survive in culture. J. Cell Biol. 1994, 126, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.A.; Kim, Y.H.; Song, Y.W. Facilitation of Fas mediated apoptosis of human chondrocytes by the proteasome inhibitor and actinomycin D. J. Rheumatol. 2003, 30, 550–558. [Google Scholar] [PubMed]
- Pelletier, J.P.; Mineau, F.; Ranger, P.; Tardif, G.; Martel-Pelletier, J. The increased synthesis of inducible nitric oxide inhibits IL-1ra synthesis by human articular chondrocytes: Possible role in osteoarthritic cartilage degradation. Osteoarthr. Cartil. 1996, 4, 77–84. [Google Scholar] [CrossRef]
- Studer, R.K.; Levicoff, E.; Georgescu, H.; Miller, L.; Jaffurs, D.; Evans, C.H. Nitric oxide inhibits chondrocyte response to IGF-I: Inhibition of IGF-IRβ tyrosine phosphorylation. Am. J. Physiol. Cell Physiol. 2000, 279, C961–C969. [Google Scholar] [PubMed]
- Del Carlo, M., Jr.; Loeser, R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheumatol. 2002, 46, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, K.; D’Lima, D.D.; Hashimoto, S.; Lotz, M. Cell death in cartilage. Osteoarthr. Cartil. 2004, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, M.; Armstrong, J.S.; Cheung, N.S.; Siau, J.L.; Rose, P.; Schantz, J.T.; Jones, D.P.; Halliwell, B. Peroxynitrite mediates calcium-dependent mitochondrial dysfunction and cell death via activation of calpains. FASEB J. 2004, 18, 1395–1397. [Google Scholar] [CrossRef] [PubMed]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; van Offel, J.F.; Bridts, C.H.; de Clerck, L.S. Influence of pro-inflammatory (IL-1α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil. 2003, 11, 681–687. [Google Scholar] [CrossRef]
- Kuhn, K.; Hashimoto, S.; Lotz, M. IL-1β protects human chondrocytes from CD95-induced apoptosis. J. Immunol. 2000, 164, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, I.; Figenschau, Y.; Olsen, E.; Bakkelund, W.; Smedsrod, B.; Sveinbjornsson, B. Tumor necrosis factor-related apoptosis-inducing ligand induces apoptosis in human articular chondrocytes in vitro. Biochem. Biophys. Res. Commun. 2002, 296, 671–676. [Google Scholar] [CrossRef]
- Lee, S.W.; Lee, H.J.; Chung, W.T.; Choi, S.M.; Rhyu, S.H.; Kim, D.K.; Kim, K.T.; Kim, J.Y.; Kim, J.M.; Yoo, Y.H. TRAIL induces apoptosis of chondrocytes and influences the pathogenesis of experimentally induced rat osteoarthritis. Arthritis Rheumatol. 2004, 50, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Sena, P.; Manfredini, G.; Benincasa, M.; Mariani, F.; Smargiassi, A.; Catani, F.; Palumbo, C. Up-regulation of the chemo-attractive receptor ChemR23 and occurrence of apoptosis in human chondrocytes isolated from fractured calcaneal osteochondral fragments. J. Anat. 2014, 224, 659–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maneiro, E.; Martin, M.A.; de Andres, M.C.; Lopez-Armada, M.J.; Fernandez-Sueiro, J.L.; del Hoyo, P.; Galdo, F.; Arenas, J.; Blanco, F.J. Mitochondrial respiratory activity is altered in osteoarthritic human articular chondrocytes. Arthritis Rheumatol. 2003, 48, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Romero, C.; Calamia, V.; Mateos, J.; Carreira, V.; Martinez-Gomariz, M.; Fernandez, M.; Blanco, F.J. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: A decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteom. 2009, 8, 172–189. [Google Scholar] [CrossRef] [PubMed]
- Grishko, V.I.; Druzhyna, N.; LeDoux, S.P.; Wilson, G.L. Nitric oxide-induced damage to mtDNA and its subsequent repair. Nucleic Acids Res. 1999, 27, 4510–4516. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Precht, P.; Balakir, R.; Horton, W.E., Jr. Evidence of a direct role for Bcl-2 in the regulation of articular chondrocyte apoptosis under the conditions of serum withdrawal and retinoic acid treatment. J. Cell. Biochem. 1998, 71, 302–309. [Google Scholar] [CrossRef]
- Engels, B.M.; Hutvagner, G. Principles and effects of microRNA-mediated post-transcriptional gene regulation. Oncogene 2006, 25, 6163–6169. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Sato, T.; Inoue, A.; Otsuki, S.; Ito, Y.; Yokoyama, S.; Kato, Y.; Takemoto, F.; Nakasa, T.; Yamashita, S.; et al. MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev. 2010, 24, 1173–1185. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheumatol. 2009, 60, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1β responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Hobert, O. Gene regulation by transcription factors and microRNAs. Science 2008, 319, 1785–1786. [Google Scholar] [CrossRef] [PubMed]
- Mobasheri, A.; Matta, C.; Zakany, R.; Musumeci, G. Chondrosenescence: Definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 2015, 80, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Pritzker, K.; Goding, J.; Terkeltaub, R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J. Rheumatol. 2001, 28, 2681–2691. [Google Scholar] [PubMed]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.D., Jr.; Loeser, R.F. Increased oxidative stress with aging reduces chondrocyte survival: Correlation with intracellular glutathione levels. Arthritis Rheumatol. 2003, 48, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Otsuki, S.; Blanco, F.J.; Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheumatol. 2010, 62, 791–801. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.; Verzijl, N.; Wenting-van Wijk, M.J.; Jacobs, K.M.; van El, B.; van Roermund, P.M.; Bank, R.A.; Bijlsma, J.W.; TeKoppele, J.M.; Lafeber, F.P. Accumulation of advanced glycation end products as a molecular mechanism for aging as a risk factor in osteoarthritis. Arthritis Rheumatol. 2004, 50, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, G.; Won, Y.; Lee, M.; Kwak, J.S.; Chun, C.H.; Chun, J.S. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 9424–9429. [Google Scholar] [CrossRef] [PubMed]
- Yamabe, S.; Hirose, J.; Uehara, Y.; Okada, T.; Okamoto, N.; Oka, K.; Taniwaki, T.; Mizuta, H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013, 280, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Guerne, P.A.; Blanco, F.; Kaelin, A.; Desgeorges, A.; Lotz, M. Growth factor responsiveness of human articular chondrocytes in aging and development. Arthritis Rheumatol. 1995, 38, 960–968. [Google Scholar] [CrossRef]
- Loeser, R.F.; Gandhi, U.; Long, D.L.; Yin, W.; Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 2014, 66, 2201–2209. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lam, G.Y.; Brumell, J.H. Autophagy signaling through reactive oxygen species. Antioxid. Redox Signal. 2011, 14, 2215–2231. [Google Scholar] [CrossRef] [PubMed]
- Vasheghani, F.; Zhang, Y.; Li, Y.H.; Blati, M.; Fahmi, H.; Lussier, B.; Roughley, P.; Lagares, D.; Endisha, H.; Saffar, B.; et al. PPARγ deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann. Rheum. Dis. 2015, 74, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Bertacchini, J.; Heidari, N.; Mediani, L.; Capitani, S.; Shahjahani, M.; Ahmadzadeh, A.; Saki, N. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell. Mol. Life Sci. 2015, 72, 2337–2247. [Google Scholar] [CrossRef] [PubMed]
- Bohensky, J.; Terkhorn, S.P.; Freeman, T.A.; Adams, C.S.; Garcia, J.A.; Shapiro, I.M.; Srinivas, V. Regulation of autophagy in human and murine cartilage: Hypoxia-inducible factor 2 suppresses chondrocyte autophagy. Arthritis Rheumatol. 2009, 60, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Knowles, M.A.; Platt, F.M.; Ross, R.L.; Hurst, C.D. Phosphatidylinositol 3-kinase (PI3K) pathway activation in bladder cancer. Cancer Metastasis Rev. 2009, 28, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Takayama, K.; Matsushita, T.; Ishida, K.; Kubo, S.; Matsumoto, T.; Fujita, N.; Oka, S.; Kurosaka, M.; Kuroda, R. Autophagy modulates osteoarthritis-related gene expression in human chondrocytes. Arthritis Rheumatol. 2012, 64, 1920–1928. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Le Toumelin, G.; Criollo, A.; Rain, J.C.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Levine, B. The autophagy effector Beclin 1: A novel BH3-only protein. Oncogene 2008, 27 (Suppl. 1), S137–S148. [Google Scholar] [CrossRef] [PubMed]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; de Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg-Lerner, A.; Bialik, S.; Simon, H.U.; Kimchi, A. Life and death partners: Apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009, 16, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Hasegawa, A.; Taniguchi, N.; Miyaki, S.; Blanco, F.J.; Lotz, M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann. Rheum. Dis. 2012, 71, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Carames, B.; Taniguchi, N.; Seino, D.; Blanco, F.J.; D’Lima, D.; Lotz, M. Mechanical injury suppresses autophagy regulators and pharmacologic activation of autophagy results in chondroprotection. Arthritis Rheumatol. 2012, 64, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Hui, W.; Young, D.A.; Rowan, A.D.; Xu, X.; Cawston, T.E.; Proctor, C.J. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Wang, W.; Zhao, Z.; Zhang, T.; Song, Y. Autophagy in human articular chondrocytes is cytoprotective following glucocorticoid stimulation. Mol. Med. Rep. 2014, 9, 2166–2172. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Figueroa, P.; Lotz, M.K.; Blanco, F.J.; Carames, B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015, 67, 966–976. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, W.; Zhang, H.; Hu, Y.; Wang, M.; Yin, Z. The dual role of autophagy in chondrocyte responses in the pathogenesis of articular cartilage degeneration in osteoarthritis. Int. J. Mol. Med. 2014, 32, 1311–1318. [Google Scholar]
- Uehara, Y.; Hirose, J.; Yamabe, S.; Okamoto, N.; Okada, T.; Oyadomari, S.; Mizuta, H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthr. Cartil. 2014, 22, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Endo, M.; Oike, Y. Endoplasmic reticulum stress-related inflammation and cardiovascular diseases. Int. J. Inflam. 2011, 2011, 259462. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Koizumi, A.; Takeda, K.; Gotoh, T.; Akira, S.; Araki, E.; Mori, M. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J. Clin. Investig. 2002, 109, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Chiang, W.C.; Kurt, T.D.; Sigurdson, C.J.; Lin, J.H. Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death. Am. J. Pathol. 2015, 185, 1800–1808. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.E.; Speicher, D.M.; Gradisar, I.; McBurney, D.L.; Baraga, A.; Doane, K.J.; Horton, W.E., Jr. Advanced osteoarthritis in humans is associated with altered collagen VI expression and upregulation of ER-stress markers Grp78 and bag-1. J. Histochem. Cytochem. 2009, 57, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Hamamura, K.; Goldring, M.B.; Yokota, H. Involvement of p38 MAPK in regulation of MMP13 mRNA in chondrocytes in response to surviving stress to endoplasmic reticulum. Arch. Oral Biol. 2009, 54, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Husa, M.; Petursson, F.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res. Ther. 2013, 15, R218. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Carlson, S.G.; McBurney, D.; Horton, W.E., Jr. Multiple signals induce endoplasmic reticulum stress in both primary and immortalized chondrocytes resulting in loss of differentiation, impaired cell growth, and apoptosis. J. Biol. Chem. 2005, 280, 31156–31165. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Romero, C.; Carreira, V.; Rego, I.; Remeseiro, S.; Lopez-Armada, M.J.; Blanco, F.J. Proteomic analysis of human osteoarthritic chondrocytes reveals protein changes in stress and glycolysis. Proteomics 2008, 8, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Albertini, M.C.; Galluzzi, L.; Buonocore, G.; Proietti, F.; Balduini, W. Increased autophagy reduces endoplasmic reticulum stress after neonatal hypoxia-ischemia: Role of protein synthesis and autophagic pathways. Exp. Neurol. 2014, 255, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Saito, S.; Sasaki, R.; Tomatsu, T.; Toyama, Y. Expression of granzyme B in human articular chondrocytes. J. Rheumatol. 2003, 30, 1799–1810. [Google Scholar] [PubMed]
- Saito, S.; Murakoshi, K.; Kotake, S.; Kamatani, N.; Tomatsu, T. Granzyme B induces apoptosis of chondrocytes with natural killer cell-like cytotoxicity in rheumatoid arthritis. J. Rheumatol. 2008, 35, 1932–1943. [Google Scholar] [PubMed]
- Wang, Q.; Rozelle, A.L.; Lepus, C.M.; Scanzello, C.R.; Song, J.J.; Larsen, D.M.; Crish, J.F.; Bebek, G.; Ritter, S.Y.; Lindstrom, T.M.; et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 2011, 17, 1674–1679. [Google Scholar] [CrossRef] [PubMed]
- Bohana-Kashtan, O.; Ziporen, L.; Donin, N.; Kraus, S.; Fishelson, Z. Cell signals transduced by complement. Mol. Immunol. 2004, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Mistry, D.; Oue, Y.; Chambers, M.G.; Kayser, M.V.; Mason, R.M. Chondrocyte death during murine osteoarthritis. Osteoarthr. Cartil. 2004, 12, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Coustry, F.; Posey, K.L.; Liu, P.; Alcorn, J.L.; Hecht, J.T. D469del-COMP retention in chondrocytes stimulates caspase-independent necroptosis. Am. J. Pathol. 2012, 180, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.Y.; Kim, H.T. Chondrocyte apoptosis induced by collagen degradation: Inhibition by caspase inhibitors and IGF-1. J. Orthop. Res. 2004, 22, 140–144. [Google Scholar] [CrossRef]
- Lopez-Armada, M.J.; Carames, B.; Cillero-Pastor, B.; Lires-Dean, M.; Maneiro, E.; Fuentes, I.; Ruiz, C.; Galdo, F.; Blanco, F.J. Phosphatase-1 and -2A inhibition modulates apoptosis in human osteoarthritis chondrocytes independently of nitric oxide production. Ann. Rheum. Dis. 2005, 64, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, M.E.; Nadeau, D.P.; Fisher, P.W.; Wang, F.; Keller, P.M.; DeWolf, W.E., Jr.; Goldring, M.B.; Badger, A.M.; Lee, D.; Levy, M.A.; et al. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J. Orthop. Res. 2000, 18, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Fernandes, J.C.; Jovanovic, D.V.; Reboul, P.; Martel-Pelletier, J. Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: Role of cyclooxygenase-2 and inducible nitric oxide synthase. J. Rheumatol. 2001, 28, 2509–2519. [Google Scholar] [PubMed]
- Loening, A.M.; James, I.E.; Levenston, M.E.; Badger, A.M.; Frank, E.H.; Kurz, B.; Nuttall, M.E.; Hung, H.H.; Blake, S.M.; Grodzinsky, A.J.; et al. Injurious mechanical compression of bovine articular cartilage induces chondrocyte apoptosis. Arch. Biochem. Biophys. 2000, 381, 205–212. [Google Scholar] [CrossRef] [PubMed]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheumatol. 2006, 54, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Lotz, M.K.; Kraus, V.B. New developments in osteoarthritis. Posttraumatic osteoarthritis: Pathogenesis and pharmacological treatment options. Arthritis Res. Ther. 2010, 12, 211. [Google Scholar] [CrossRef] [PubMed]
- Elsaid, K.A.; Zhang, L.; Shaman, Z.; Patel, C.; Schmidt, T.A.; Jay, G.D. The impact of early intra-articular administration of interleukin-1 receptor antagonist on lubricin metabolism and cartilage degeneration in an anterior cruciate ligament transection model. Osteoarthr. Cartil. 2015, 23, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Mineau, F.; Boileau, C.; Martel-Pelletier, J. Diacerein reduces the level of cartilage chondrocyte DNA fragmentation and death in experimental dog osteoarthritic cartilage at the same time that it inhibits caspase-3 and inducible nitric oxide synthase. Clin. Exp. Rheumatol. 2003, 21, 171–177. [Google Scholar] [PubMed]
- Pelletier, J.P.; Yaron, M.; Haraoui, B.; Cohen, P.; Nahir, M.A.; Choquette, D.; Wigler, I.; Rosner, I.A.; Beaulieu, A.D. Efficacy and safety of diacerein in osteoarthritis of the knee: A double-blind, placebo-controlled trial. The Diacerein Study Group. Arthritis Rheumatol. 2000, 43, 2339–2348. [Google Scholar] [CrossRef]
- Takahashi, K.; Hashimoto, S.; Kubo, T.; Hirasawa, Y.; Lotz, M.; Amiel, D. Effect of hyaluronan on chondrocyte apoptosis and nitric oxide production in experimentally induced osteoarthritis. J. Rheumatol. 2000, 27, 1713–1720. [Google Scholar] [PubMed]
- De Mattei, M.; Pellati, A.; Pasello, M.; de Terlizzi, F.; Massari, L.; Gemmati, D.; Caruso, A. High doses of glucosamine-HCl have detrimental effects on bovine articular cartilage explants cultured in vitro. Osteoarthr. Cartil. 2002, 10, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Robson Brown, K.; Tarlton, J.F.; Adams, M.A.; Torlot, G.E.; Cartwright, C.; Cook, W.A.; Vassilevskaja, K.; Sharif, M. Subchondral bone plate thickening precedes chondrocyte apoptosis and cartilage degradation in spontaneous animal models of osteoarthritis. Biomed Res. Int. 2014, 2014, 606870. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035-26054. https://doi.org/10.3390/ijms161125943
Hwang HS, Kim HA. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. International Journal of Molecular Sciences. 2015; 16(11):26035-26054. https://doi.org/10.3390/ijms161125943
Chicago/Turabian StyleHwang, Hyun Sook, and Hyun Ah Kim. 2015. "Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis" International Journal of Molecular Sciences 16, no. 11: 26035-26054. https://doi.org/10.3390/ijms161125943
APA StyleHwang, H. S., & Kim, H. A. (2015). Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. International Journal of Molecular Sciences, 16(11), 26035-26054. https://doi.org/10.3390/ijms161125943