
Roles of Sestrin2 and Ribosomal Protein S6 in Transient Global Ischemia-Induced Hippocampal Neuronal Injury

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
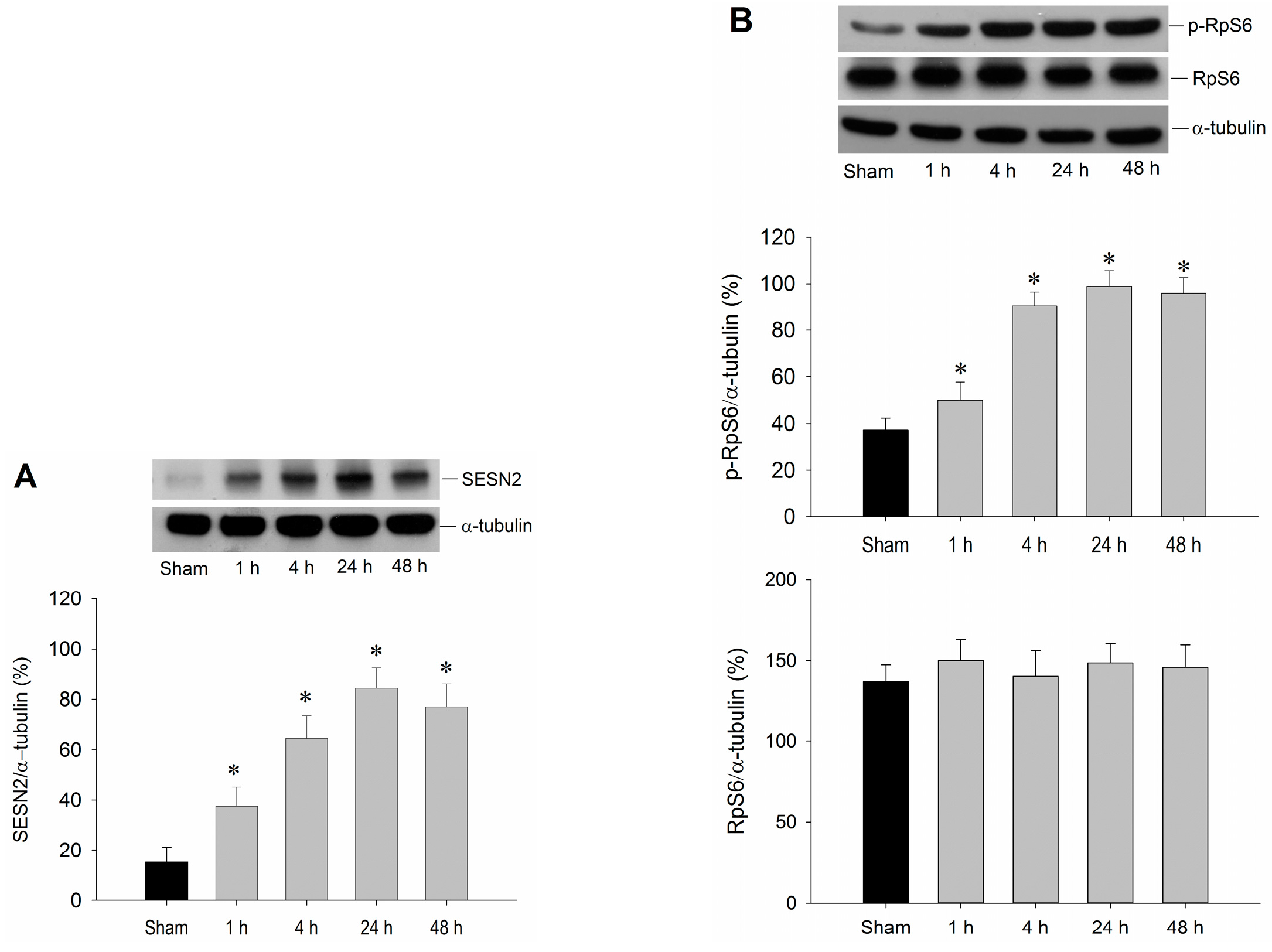
2.1. Temporal Changes of Sestrin2 and RpS6 Expressions in the Hippocampal CA1 Subfield after TGI
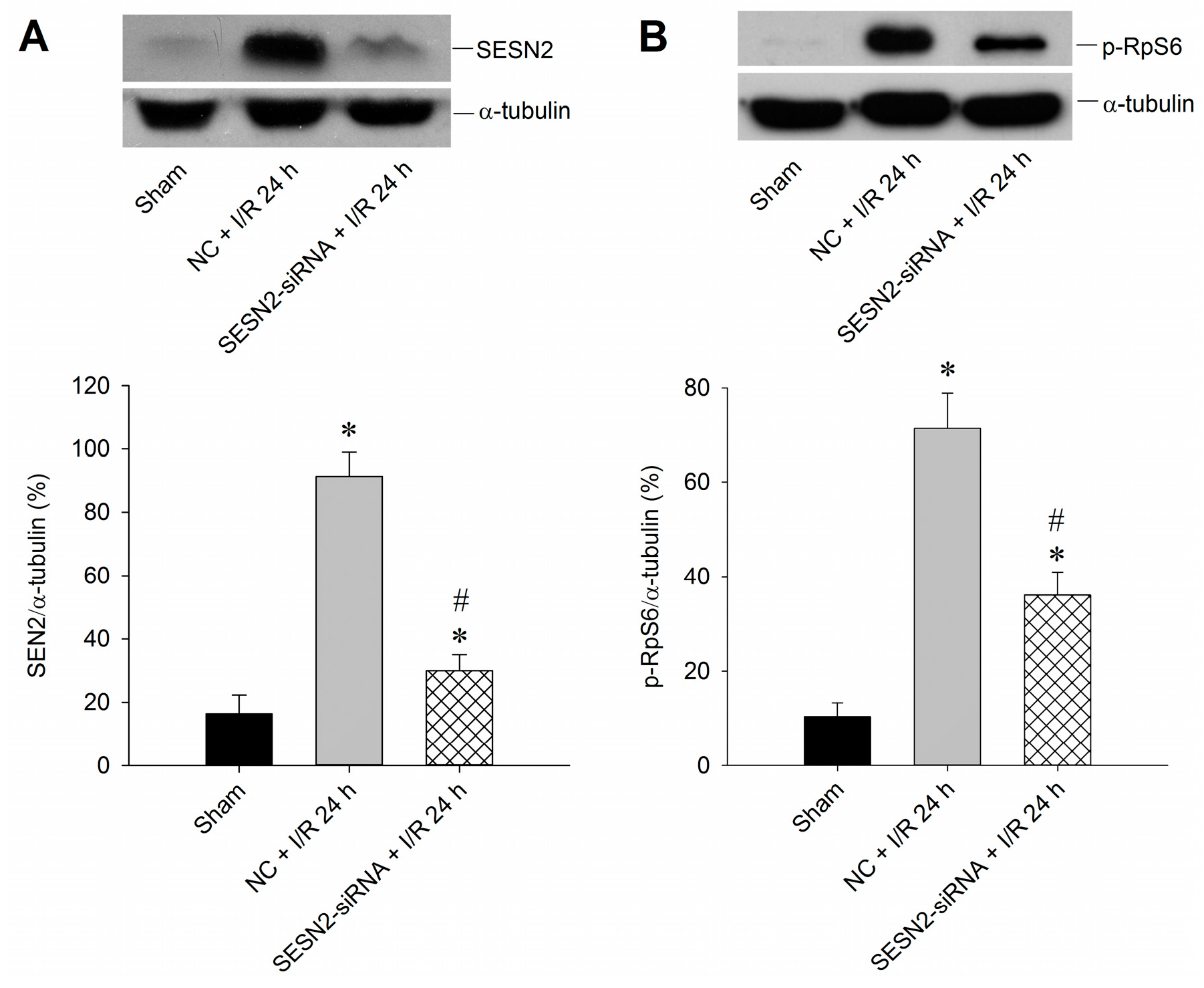
2.2. Sestrin2 siRNA Silences Sestrin2 Expression and Diminishes p-RpS6 Expression in the Hippocampal CA1 Subfield after TGI


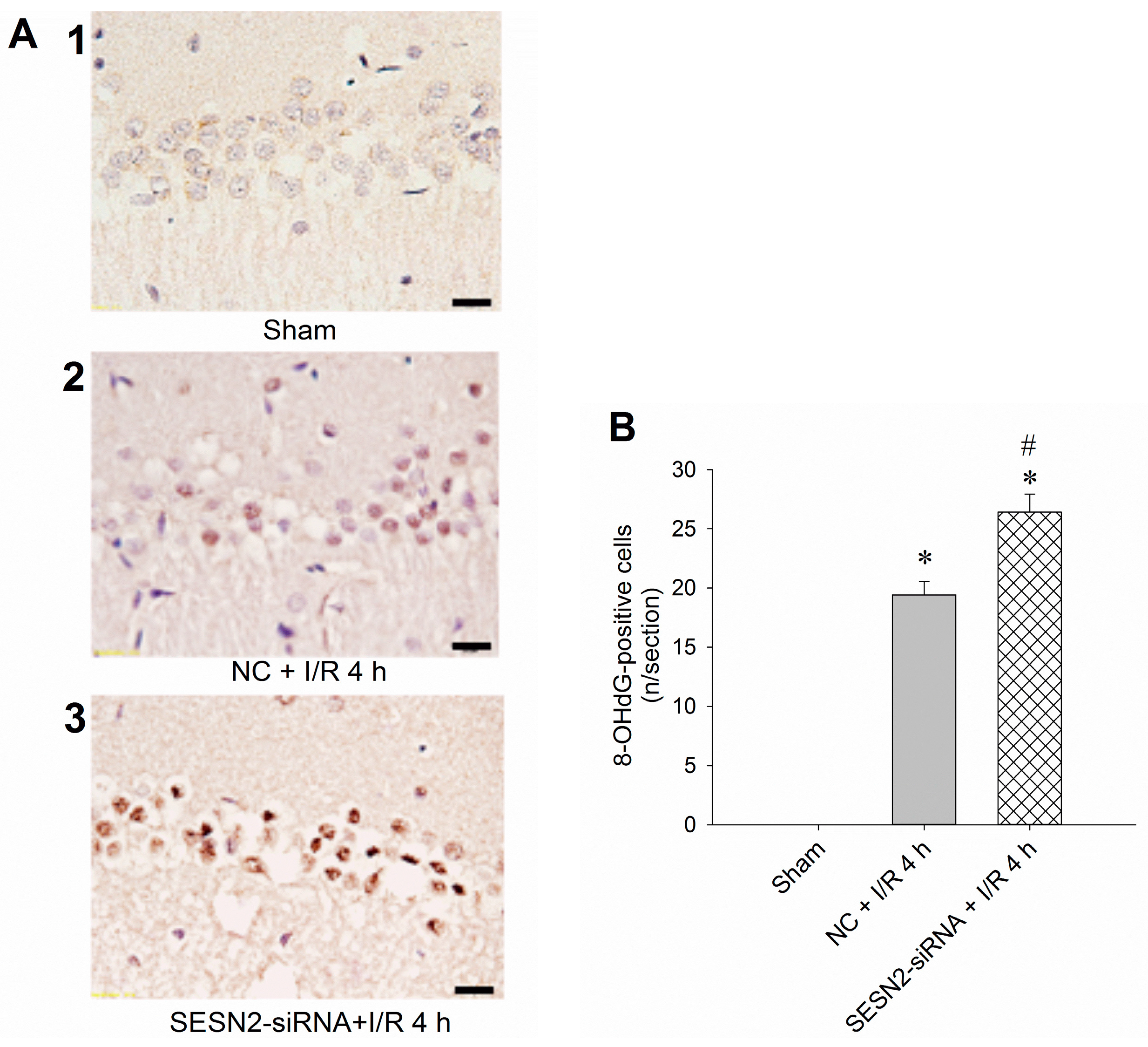
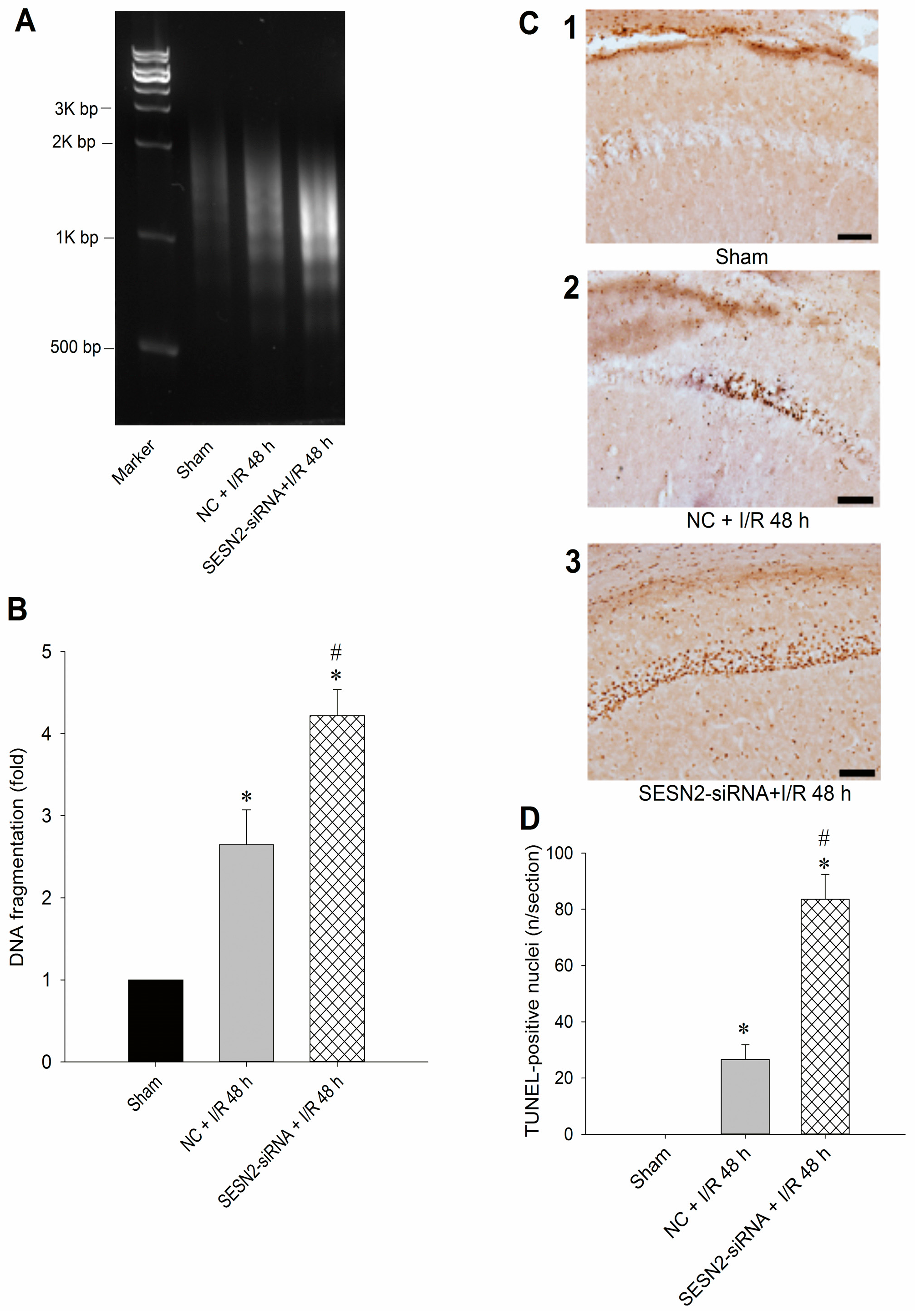
2.3. Injection of Sestrin2-siRNA Augments Oxidative Stress and Increases Neuronal Injury in the Hippocampal CA1 Subfield after TGI

2.4. Discussion

3. Experimental Section
3.1. Animals and Transient Global Ischemia
3.2. siRNA Administration
3.3. Tissue Sample Collection from the Hippocampus
3.4. Western Blotting
3.5. Immunohistochemical Staining
3.6. Qualitative and Quantitative Analysis of DNA Fragmentation
3.7. Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling (TUNEL) Staining
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Budanov, A.V.; Shoshani, T.; Faerman, A.; Zelin, E.; Kamer, I.; Kalinski, H.; Gorodin, S.; Fishman, A.; Chajut, A.; Einat, P.; et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene 2002, 21, 6017–6031. [Google Scholar] [CrossRef] [PubMed]
- Peeters, H.; Debeer, P.; Bairoch, A.; Wilquet, V.; Huysmans, C.; Parthoens, E.; Fryns, J.P.; Gewillig, M.; Nakamura, Y.; Niikawa, N.; et al. PA26 is a candidate gene for heterotaxia in humans: Identification of a novel PA26-related gene family in human and mouse. Hum. Genet. 2003, 112, 573–580. [Google Scholar] [PubMed]
- Budanov, A.V.; Sablina, A.A.; Feinstein, E.; Koonin, E.V.; Chumakov, P.M. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science 2004, 304, 596–600. [Google Scholar] [CrossRef]
- Velasco-Miguel, S.; Buckbinder, L.; Jean, P.; Gelbert, L.; Talbott, R.; Laidlaw, J.; Seizinger, B.; Kley, N. PA26, a novel target of the p53 tumor suppressor and member of the GADD family of DNA damage and growth arrest inducible genes. Oncogene 1999, 18, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxid. Redox Signal. 2011, 15, 1679–1690. [Google Scholar] [CrossRef] [PubMed]
- Essler, S.; Dehne, N.; Brune, B. Role of sestrin2 in peroxide signaling in macrophages. FEBS Lett. 2009, 583, 3531–3535. [Google Scholar] [CrossRef] [PubMed]
- Sanli, T.; Linher-Melville, K.; Tsakiridis, T.; Singh, G. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS ONE 2012, 7, e32035. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.Y.; Jin, S.H.; Cho, I.J.; Ki, S.H. Nrf2-ARE pathway regulates induction of Sestrin-2 expression. Free Radic. Biol. Med. 2012, 53, 834–841. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Chen, S.D.; Wu, C.L.; Huang, S.S.; Yang, D.I. Induction of sestrin2 as an endogenous protective mechanism against amyloid beta-peptide neurotoxicity in primary cortical culture. Exp. Neurol. 2014, 253, 63–71. [Google Scholar] [CrossRef]
- Wu, C.L.; Chen, S.D.; Yin, J.H.; Hwang, C.S.; Yang, D.I. Nuclear Factor-kappaB-dependent sestrin2 induction mediates the antioxidant effects of BDNF against mitochondrial inhibition in rat cortical neurons. Mol. Neurobiol. 2015, 26. [Google Scholar] [CrossRef]
- Meyuhas, O. Physiological roles of ribosomal protein S6: One of its kind. Int. Rev. Cell Mol. Biol. 2008, 268, 1–37. [Google Scholar] [PubMed]
- Sulic, S.; Panic, L.; Barkic, M.; Mercep, M.; Uzelac, M.; Volarevic, S. Inactivation of S6 ribosomal protein gene in T lymphocytes activates a p53-dependent checkpoint response. Genes Dev. 2005, 19, 3070–3082. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ferlito, M.; Aponte, A.; Kuno, A.; Miura, T.; Murphy, E.; Steenbergen, C. Pivotal role of mTORC2 and involvement of ribosomal protein S6 in cardioprotective signaling. Circ. Res. 2014, 114, 1268–1280. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Solhaug, A.; Torgersen, M.L.; Holme, J.A.; Lagadic-Gossmann, D.; Eriksen, G.S. Autophagy and senescence, stress responses induced by the DNA-damaging mycotoxin alternariol. Toxicology 2014, 326, 119–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2012, 441, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of PTEN-induced putative kinase 1 and dynamin-related protein 1 in transient global ischemia-induced hippocampal neuronal injury. Biochem. Biophys. Res. Commun. 2015, 460, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Lin, T.K.; Lin, J.W.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Activation of calcium/calmodulin-dependent protein kinase IV and peroxisome proliferator-activated receptor gamma coactivator-1alpha signaling pathway protects against neuronal injury and promotes mitochondrial biogenesis in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 3144–3154. [Google Scholar] [PubMed]
- Chen, S.D.; Lin, T.K.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Protective effects of peroxisome proliferator-activated receptors gamma coactivator-1alpha against neuronal cell death in the hippocampal CA1 subfield after transient global ischemia. J. Neurosci. Res. 2010, 88, 605–613. [Google Scholar] [PubMed]
- Sugawara, T.; Kawase, M.; Lewen, A.; Noshita, N.; Gasche, Y.; Fujimura, M.; Chan, P.H. Effect of hypotension severity on hippocampal CA1 neurons in a rat global ischemia model. Brain Res. 2000, 877, 281–287. [Google Scholar] [CrossRef]
- Chen, S.D.; Wu, H.Y.; Yang, D.I.; Lee, S.Y.; Shaw, F.Z.; Lin, T.K.; Liou, C.W.; Chuang, Y.C. Effects of rosiglitazone on global ischemia-induced hippocampal injury and expression of mitochondrial uncoupling protein 2. Biochem. Biophys. Res. Commun. 2006, 351, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.D.; Lee, J.M.; Yang, D.I.; Nassief, A.; Hsu, C.Y. Combination therapy for ischemic stroke: Potential of neuroprotectants plus thrombolytics. Am. J. Cardiovasc. Drugs 2002, 2, 303–313. [Google Scholar] [CrossRef]
- Chen, S.D.; Yang, D.I.; Lin, T.K.; Shaw, F.Z.; Liou, C.W.; Chuang, Y.C. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 7199–7215. [Google Scholar] [CrossRef]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97–107. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Liu, L.; Zhang, S.; Nanda, A.; Li, G. Role of inflammation and its mediators in acute ischemic stroke. J. Cardiovasc. Transl. Res. 2013, 6, 834–851. [Google Scholar] [CrossRef]
- Niizuma, K.; Yoshioka, H.; Chen, H.; Kim, G.S.; Jung, J.E.; Katsu, M.; Okami, N.; Chan, P.H. Mitochondrial and apoptotic neuronal death signaling pathways in cerebral ischemia. Biochim. Biophys. Acta 2010, 1802, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Minnerup, J.; Sutherland, B.A.; Buchan, A.M.; Kleinschnitz, C. Neuroprotection for stroke: Current status and future perspectives. Int. J. Mol. Sci. 2012, 13, 11753–11772. [Google Scholar] [CrossRef] [PubMed]
- George, P.M.; Steinberg, G.K. Novel stroke therapeutics: Unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol. Neurobiol. 2010, 41, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Dirat, B.; Laurent, K.; Puissant, A.; Auberger, P.; Budanov, A.; Tanti, J.F.; Bost, F. Sestrin2 integrates Akt and mTOR signaling to protect cells against energetic stress-induced death. Cell Death Differ. 2013, 20, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Budanov, A.V.; Talukdar, S.; Park, E.J.; Park, H.L.; Park, H.W.; Bandyopadhyay, G.; Li, N.; Aghajan, M.; Jang, I.; et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell Metab. 2012, 16, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Urushido, M.; Hamada, K.; Matsumoto, T.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Horino, T.; Fujieda, M.; et al. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am. J. Physiol. Ren. Physiol. 2013, 305, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.; Chen, L.; Wang, J.; Zhang, M.; Yang, H.; Ma, Y.; Budanov, A.; Lee, J.H.; Karin, M.; Li, J. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015, 29, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Jonassen, A.K.; Sack, M.N.; Mjos, O.D.; Yellon, D.M. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001, 89, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Salloum, F.; Das, A.; Xi, L.; Vetrovec, G.W.; Kukreja, R.C. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J. Mol. Cell. Cardiol. 2006, 41, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Volpe, M.; Sadoshima, J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ. Res. 2014, 114, 549–564. [Google Scholar] [CrossRef]
- Chuang, Y.C.; Lin, T.K.; Huang, H.Y.; Chang, W.N.; Liou, C.W.; Chen, S.D.; Chang, A.Y.; Chan, S.H. Peroxisome proliferator-activated receptors gamma/mitochondrial uncoupling protein 2 signaling protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus. J. Neuroinflamm. 2012, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Wu, C.A.; Wu, C.W.; Chan, S.H.; Chang, A.Y.; Chan, J.Y. Redox-sensitive oxidation and phosphorylation of PTEN contribute to enhanced activation of PI3K/Akt signaling in rostral ventrolateral medulla and neurogenic hypertension in spontaneously hypertensive rats. Antioxid. Redox Signal. 2013, 18, 36–50. [Google Scholar] [CrossRef]
- Chang, C.C.; Chen, S.D.; Lin, T.K.; Chang, W.N.; Liou, C.W.; Chang, A.Y.; Chan, S.H.; Chuang, Y.C. Heat shock protein 70 protects against seizure-induced neuronal cell death in the hippocampus following experimental status epilepticus via inhibition of nuclear factor-kappaB activation-induced nitric oxide synthase II expression. Neurobiol. Dis. 2014, 62, 241–249. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chuang, Y.-C.; Yang, J.-L.; Yang, D.-I.; Lin, T.-K.; Liou, C.-W.; Chen, S.-D. Roles of Sestrin2 and Ribosomal Protein S6 in Transient Global Ischemia-Induced Hippocampal Neuronal Injury. Int. J. Mol. Sci. 2015, 16, 26406-26416. https://doi.org/10.3390/ijms161125963
Chuang Y-C, Yang J-L, Yang D-I, Lin T-K, Liou C-W, Chen S-D. Roles of Sestrin2 and Ribosomal Protein S6 in Transient Global Ischemia-Induced Hippocampal Neuronal Injury. International Journal of Molecular Sciences. 2015; 16(11):26406-26416. https://doi.org/10.3390/ijms161125963
Chicago/Turabian StyleChuang, Yao-Chung, Jenq-Lin Yang, Ding-I Yang, Tsu-Kung Lin, Chia-Wei Liou, and Shang-Der Chen. 2015. "Roles of Sestrin2 and Ribosomal Protein S6 in Transient Global Ischemia-Induced Hippocampal Neuronal Injury" International Journal of Molecular Sciences 16, no. 11: 26406-26416. https://doi.org/10.3390/ijms161125963
APA StyleChuang, Y. -C., Yang, J. -L., Yang, D. -I., Lin, T. -K., Liou, C. -W., & Chen, S. -D. (2015). Roles of Sestrin2 and Ribosomal Protein S6 in Transient Global Ischemia-Induced Hippocampal Neuronal Injury. International Journal of Molecular Sciences, 16(11), 26406-26416. https://doi.org/10.3390/ijms161125963