
Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy



Abstract
:1. Introduction
2. IR-Induced Cell Death Outcomes

2.1. Apoptosis
2.2. Necrosis
2.3. Autophagy
2.4. Mitotic Catastrophe
2.5. Programmed Necrosis
2.6. Senescence
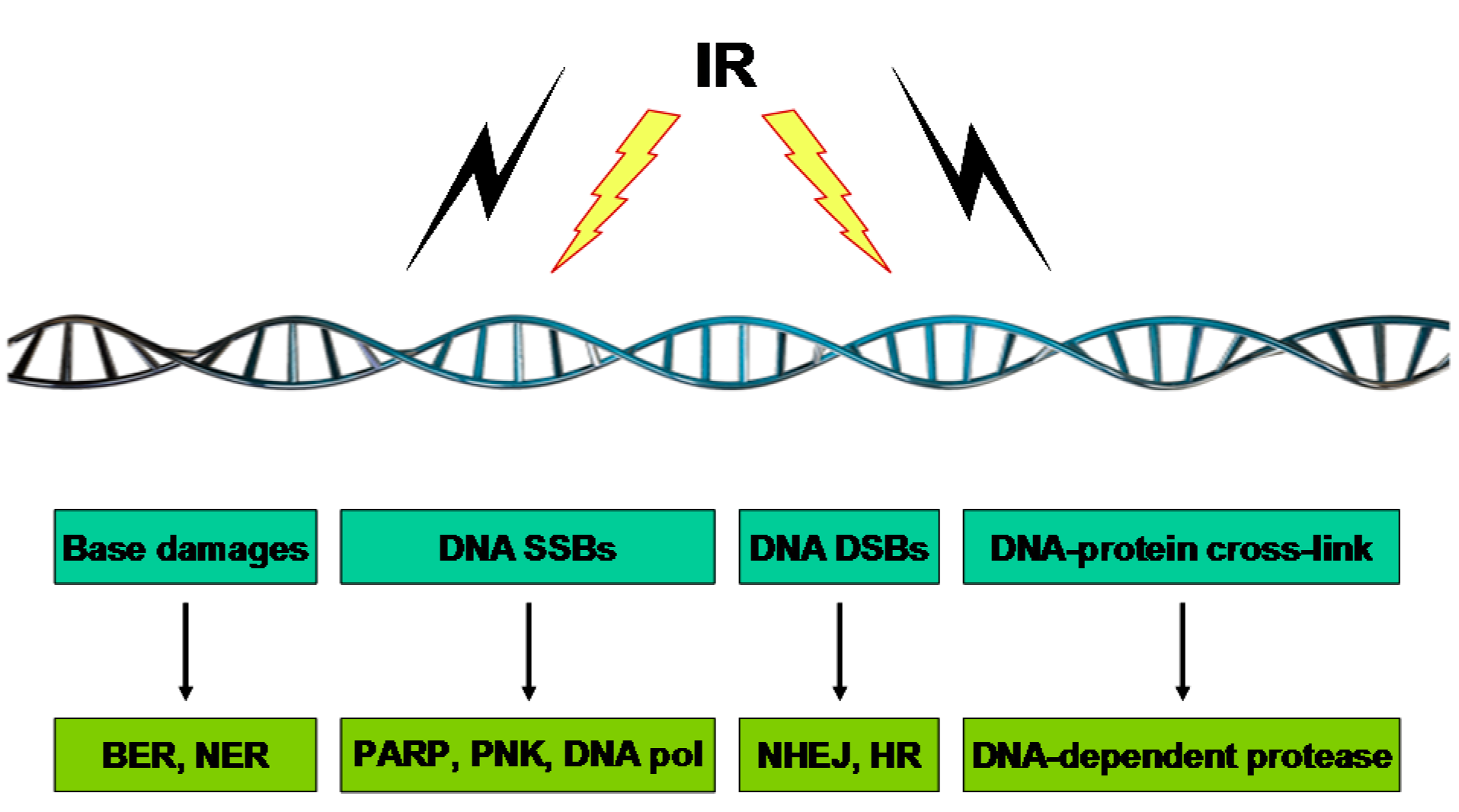
3. Genomic Instability and Cytogenetic Alterations Induced by IR
3.1. Base Damage

3.2. DNA SSBs
3.3. DNA DSBs
3.4. DNA–Protein Crosslinks
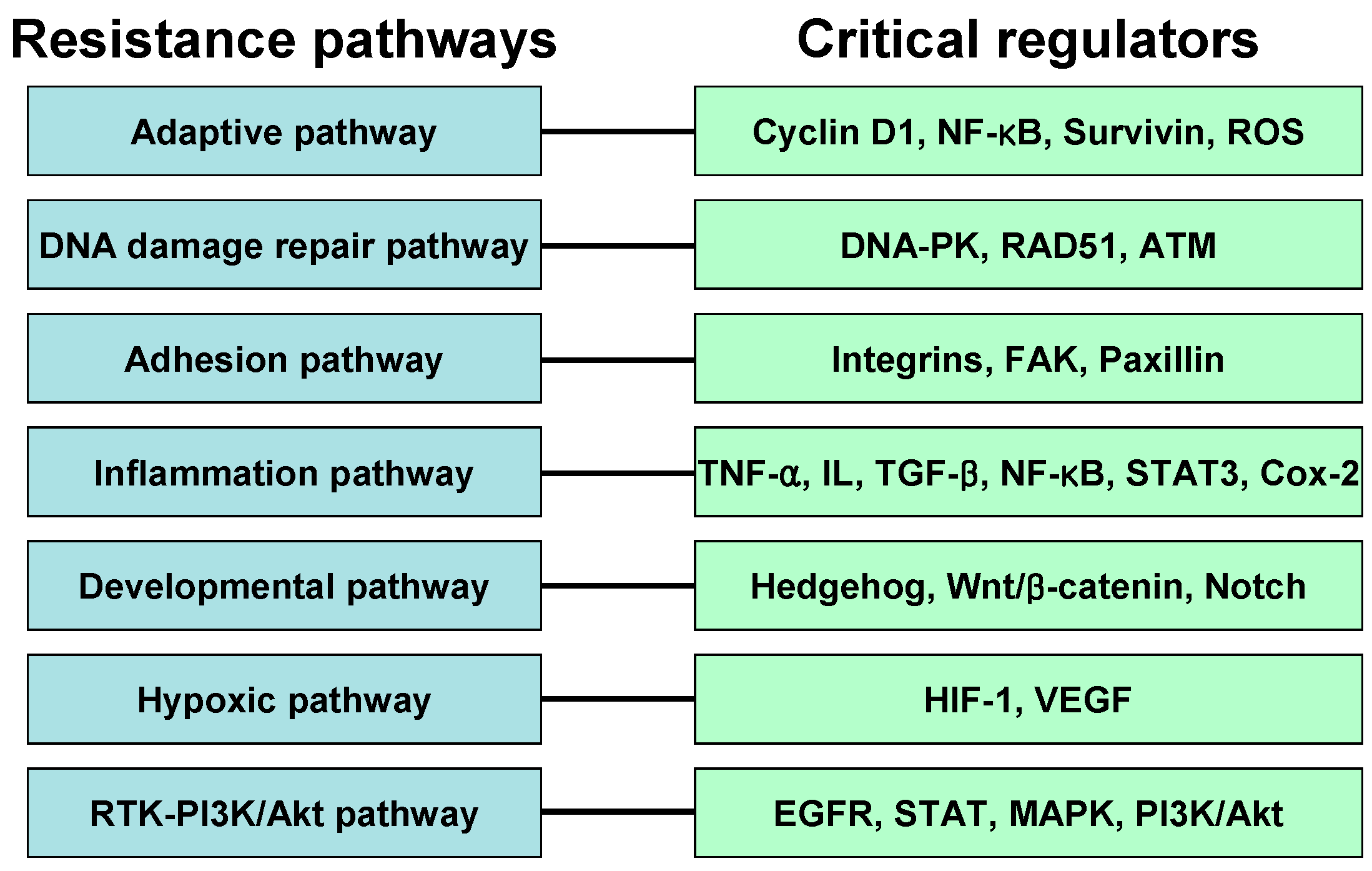
4. Resistance Pathway

4.1. Adaptive Pathway
4.2. DNA Damage Repair Pathway
4.3. Adhesion Pathway
4.4. Inflammation Pathway
4.5. Developmental Pathway
4.6. Hypoxia Pathway
4.7. RTK-PI3K-Akt Pathway
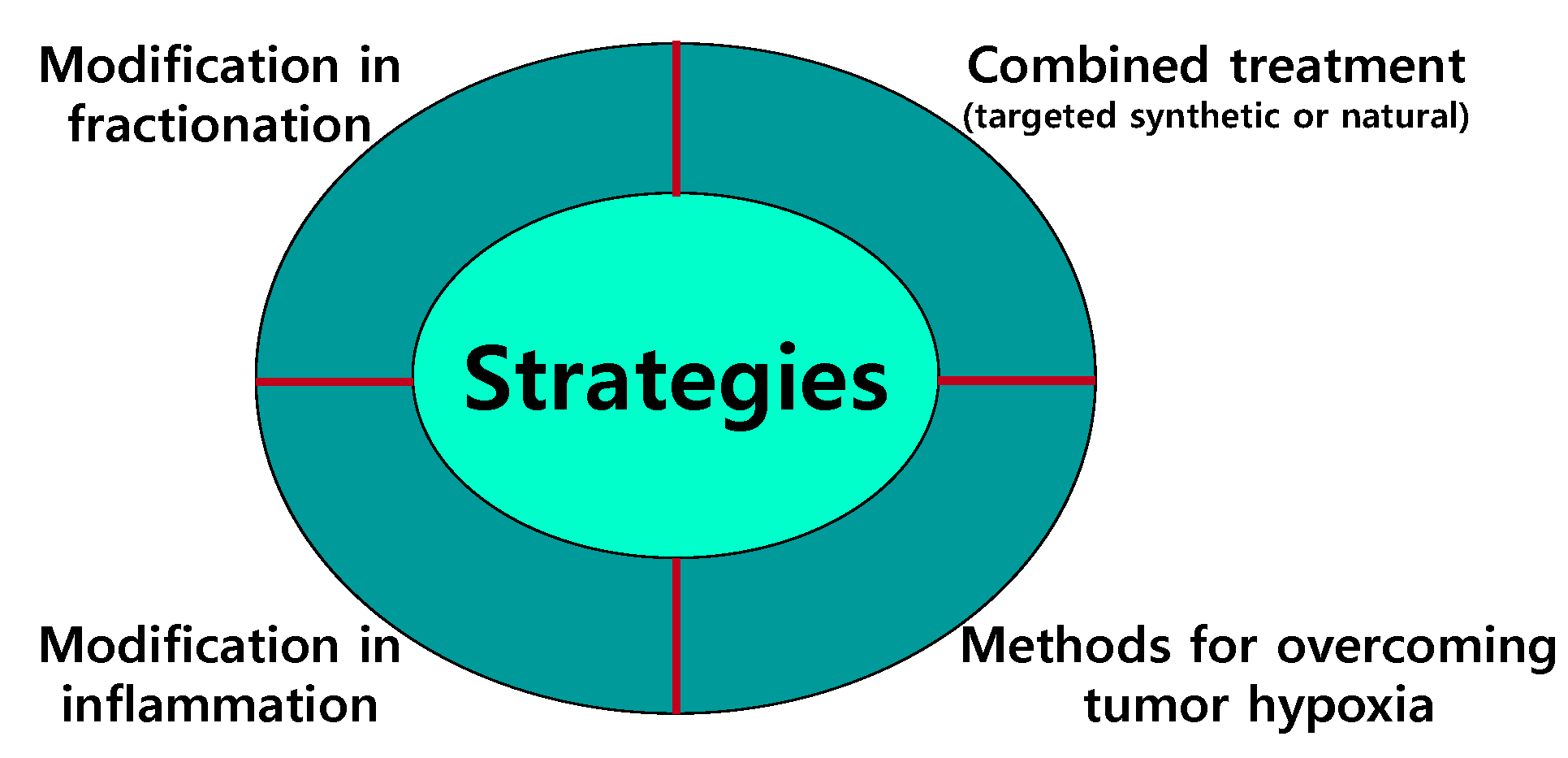
5. Strategies for Overcoming Resistance to IR

5.1. Modifications in Fractionation
5.2. Combined Treatment
5.2.1. Selective Molecularly Targeted Synthetic Agents
5.2.2. Radiosensitizers Derived from Natural Products
Curcumin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Types | Source | Radiosensitization | Target in Radiosensitization | Radioprotection | Target in Radioprotection |
|---|---|---|---|---|---|---|
| Curcumin | Polyphenol | Tumeric | Arrest, Apoptosis | NF-kB | Yes | NRF2, Antioxidant enzymes |
| Resveratrol | Polyphenol | Grapes | Apoptosis, Senescence | NF-kB, Cox-2, 5-LOX | Yes | Not determined |
| Genistein | Polyphenol | Soybean | Arrest, Apoptosis | Akt, Erk, Survivin, Cycline B, NF-kB | Yes | Not determined |
| Quercetin | Polyphenol | Ubiquitous | Arrest, Apoptosis | ATM | Yes | Not determined |
Resveratrol
Genistein
Quercetin
5.3. Modification of Inflammation
5.4. Methods of Overcoming Tumor Hypoxia
6. Perspectives and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hall, E.J. Cancer caused by X-rays—A random event? Lancet Oncol. 2007, 8, 369–370. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. GENOMIC INSTABILITY IN CANCER Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Kolesnick, R.; Fuks, Z. Radiation and ceramide-induced apoptosis. Oncogene 2003, 22, 5897–5906. [Google Scholar] [CrossRef] [PubMed]
- D'Amours, D.; Desnoyers, S.; D'Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Simbulan-Rosenthal, C.M.; Rosenthal, D.S.; Iyer, S.; Boulares, A.H.; Smulson, M.E. Transient poly(ADP-ribosyl)ation of nuclear proteins and role of poly(ADP-ribose) polymerase in the early stages of apoptosis. J. Biol. Chem. 1998, 273, 13703–13712. [Google Scholar] [CrossRef] [PubMed]
- Kelland, L.R.; Tonkin, K.S. The effect of 3-aminobenzamide in the radiation response of 3 human cervix carcinoma xenografts. Radiother. Oncol. 1989, 15, 363–369. [Google Scholar] [CrossRef]
- Calabrese, C.R.; Almassy, R.; Barton, S.; Batey, M.A.; Calvert, A.H.; Canan-Koch, S.; Durkacz, B.W.; Hostomsky, Z.; Kumpf, R.A.; Kyle, S.; et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J. Natl. Cancer Inst. 2004, 96, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Donawho, C.K.; Luo, Y.; Luo, Y.; Penning, T.D.; Bauch, J.L.; Bouska, J.J.; Bontcheva-Diaz, V.D.; Cox, B.F.; DeWeese, T.L.; Dillehay, L.E.; et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin. Cancer Res. 2007, 13, 2728–2737. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.; Araki, K.; Wang, D.Y.; Li, G.Y.; Li, X.; Zhang, J.; Xu, W.Z.; Hoover, R.K.; Lauter, S.; O'Malley, B.; et al. Head and neck cancer radiosensitization by the novel poly(ADP-ribose) polymerase inhibitor GPI-15427. Head Neck 2010, 32, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Senra, J.M.; Telfer, B.A.; Cherry, K.E.; McCrudden, C.M.; Hirst, D.G.; O'Connor, M.J.; Wedge, S.R.; Stratford, I.J. Inhibition of PARP-1 by olaparib (AZD2281) increases the radiosensitivity of a lung tumor xenograft. Mol. Cancer Ther. 2011, 10, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.W.; Chen, B.P.C.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002, 16, 2333–2338. [Google Scholar] [CrossRef] [PubMed]
- Shrivastav, M.; Miller, C.A.; de Haro, L.P.; Durant, S.T.; Chen, B.P.C.; Chen, D.J.; Nickoloff, J.A. DNA-PKcs and ATM co-regulate DNA double-strand break repair. DNA Repair 2009, 8, 920–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, J.Q.; Cao, X.F.; Zhang, Q.M.; Lim, C.U.K.; Ullrich, R.L.; Bailey, S.M.; Liber, H.L. Partial deficiency of DNA-PKcs increases ionizing radiation-induced mutagenesis and telomere instability in human cells. Cancer Lett. 2007, 250, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett. 2001, 490, 117–122. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Dou, Q.P. Putative roles of retinoblastoma protein in apoptosis. Apoptosis 1997, 2, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Delston, R.B.; Harbour, J.W. Rb at the interface between cell cycle and apoptotic decisions. Curr. Mol. Med. 2006, 6, 713–718. [Google Scholar] [PubMed]
- An, B.; Dou, Q.P. Cleavage of retinoblastoma protein during apoptosis: An interleukin 1 beta-converting enzyme-like protease as candidate. Cancer Res. 1996, 56, 438–442. [Google Scholar] [PubMed]
- Watters, D. Molecular mechanisms of ionizing radiation-induced apoptosis. Immunol. Cell Biol. 1999, 77, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, T.; Reed, J.C. Tumor-suppressor P53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [PubMed]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; Walls, K.C.; Ghosh, A.P.; Akhtar, R.S.; Klocke, B.J.; Roth, K.A. Cytoplasmic p53 and activated bax regulate p53-dependent, transcription-independent neural precursor cell apoptosis. J. Histochem. Cytochem. 2010, 58, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.A.; Dazin, P.; Hu, L.; Deen, D.F.; Israel, A. P53-independent apoptosis: A mechanism of radiation-induced cell death of glioblastoma cells. Cancer J. Sci. Am. 1996, 2, 114–121. [Google Scholar] [PubMed]
- Kyprianou, N.; Rock, S. Radiation-induced apoptosis of human prostate cancer cells is independent of mutant p53 overexpression. Anticancer Res. 1998, 18, 897–905. [Google Scholar] [PubMed]
- Bracey, T.S.; Miller, J.C.; Preece, A.; Paraskeva, C. Gamma-radiation-induced apoptosis in human colorectal adenoma and carcinoma cell lines can occur in the absence of wild type p53. Oncogene 1995, 10, 2391–2396. [Google Scholar] [PubMed]
- Barlow, C.; Brown, K.D.; Deng, C.X.; Tagle, D.A.; Wynshaw-Boris, A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 1997, 17, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Reinke, V.; Lozano, G. The p53 targets mdm2 and Fas are not required as mediators of apoptosis in vivo. Oncogene 1997, 15, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Chmura, S.J.; Nodzenski, E.; Beckett, M.A.; Kufe, D.W.; Quintans, J.; Weichselbaum, R.R. Loss of ceramide production confers resistance to radiation-induced apoptosis. Cancer Res. 1997, 57, 1270–1275. [Google Scholar] [PubMed]
- Michael, J.M.; Lavin, M.F.; Watters, D.J. Resistance to radiation-induced apoptosis in Burkitt's lymphoma cells is associated with defective ceramide signaling. Cancer Res. 1997, 57, 3600–3605. [Google Scholar] [PubMed]
- Basu, S.; Bayoumy, S.; Zhang, Y.; Lozano, J.; Kolesnick, R. BAD enables ceramide to signal apoptosis via Ras and Raf-1. J. Biol. Chem. 1998, 273, 30419–30426. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, W.D.; Fornari, F.A., Jr.; Browning, J.L.; Gewirtz, D.A.; Kolesnick, R.N.; Grant, S. Attenuation of ceramide-induced apoptosis by diglyceride in human myeloid leukemia cells. J. Biol. Chem. 1994, 269, 31685–31692. [Google Scholar] [PubMed]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol. Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [PubMed]
- Herr, I.; Wilhelm, D.; Bohler, T.; Angel, P.; Debatin, K.M. Activation of CD95 (APO-1/Fas) signaling by ceramide mediates cancer therapy-induced apoptosis. EMBO J. 1997, 16, 6200–6208. [Google Scholar] [CrossRef] [PubMed]
- Rainaldi, G.; Ferrante, A.; Indovina, P.L.; Santini, M.T. Induction of apoptosis or necrosis by ionizing radiation is dose-dependent in MG-63 osteosarcoma multicellular spheroids. Anticancer Res. 2003, 23, 2505–2518. [Google Scholar] [PubMed]
- Takeuchi, H.; Kondo, Y.; Fujiwara, K.; Kanzawa, T.; Aoki, H.; Mills, G.B.; Kondo, S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005, 65, 3336–3346. [Google Scholar] [PubMed]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Zhuang, W.; Qin, Z.; Liang, Z. The role of autophagy in sensitizing malignant glioma cells to radiation therapy. Acta Biochim. Biophys. Sin. 2009, 41, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Liu, Y.W.; Wang, Y.K.; Lin, T.H.; Li, Y.Z.; Chen, S.H.; Lee, Y.R. Ionizing radiation induces autophagy in human oral squamous cell carcinoma. J. Balk. Union Oncol. 2014, 19, 137–144. [Google Scholar]
- Jo, G.H.; Bogler, O.; Chwae, Y.J.; Yoo, H.; Lee, S.H.; Park, J.B.; Kim, Y.J.; Kim, J.H.; Gwak, H.S. Radiation-induced autophagy contributes to cell death and induces apoptosis partly in malignant glioma cells. Cancer Res. Treat. 2015, 47, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Du, J.; Hua, S.; Zhang, H.; Gu, C.; Wang, J.; Yang, L.; Huang, J.; Yu, J.; Liu, F. Suppression of autophagy augments the radiosensitizing effects of STAT3 inhibition on human glioma cells. Exp. Cell Res. 2015, 330, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Vares, G.; Ory, K.; Lectard, B.; Levalois, C.; Altmeyer-Morel, S.; Chevillard, S.; Lebeau, J. Progesterone prevents radiation-induced apoptosis in breast cancer cells. Oncogene 2004, 23, 4603–4613. [Google Scholar] [CrossRef] [PubMed]
- Luce, A.; Courtin, A.; Levalois, C.; Altmeyer-Morel, S.; Romeo, P.H.; Chevillard, S.; Lebeau, J. Death receptor pathways mediate targeted and non-targeted effects of ionizing radiations in breast cancer cells. Carcinogenesis 2009, 30, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Ianzini, F.; Bertoldo, A.; Kosmacek, E.A.; Phillips, S.L.; Mackey, M.A. Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int. 2006, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.A.; Hermeking, H.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B. 14–3-3Sigma is required to prevent mitotic catastrophe after DNA damage. Nature 1999, 401, 616–620. [Google Scholar] [PubMed]
- Ianzini, F.; Mackey, M.A. Spontaneous premature chromosome condensation and mitotic catastrophe following irradiation of HeLa S3 cells. Int. J. Radiat. Biol. 1997, 72, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Ree, A.H.; Stokke, T.; Bratland, A.; Patzke, S.; Nome, R.V.; Folkvord, S.; Meza-Zepeda, L.A.; Flatmark, K.; Fodstad, O.; Andersson, Y. DNA damage responses in cell cycle G2 phase and mitosis—Tracking and targeting. Anticancer Res. 2006, 26, 1909–1916. [Google Scholar] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1–RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Nehs, M.A.; Lin, C.I.; Kozono, D.E.; Whang, E.E.; Cho, N.L.; Zhu, K.; Moalem, J.; Moore, F.D., Jr.; Ruan, D.T. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery 2011, 150, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Le, O.N.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdiere, C.; Kokta, V.; Haddad, E.; Beausejour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.S.; Yu, Y.C.; Lee, Y.J.; Chen, J.H.; Hsu, H.Y.; Chiu, S.J. Depletion of securin induces senescence after irradiation and enhances radiosensitivity in human cancer cells regardless of functional p53 expression. Int. J. Radiat. Oncol. Biol. Phys. 2010, 77, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Scott, A.; Cameron, M.; Murray, D. Induction of accelerated senescence by gamma radiation in human solid tumor-derived cell lines expressing wild-type TP53. Radiat. Res. 2005, 163, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Yount, C.; Lang, H.; Yang, A.; Riemer, E.C.; Lyons, K.; Vanek, K.N.; Silvestri, G.A.; Schulte, B.A.; Wang, G.Y. Activation of p53 with Nutlin-3a radiosensitizes lung cancer cells via enhancing radiation-induced premature senescence. Lung Cancer 2013, 81, 167–73. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Boothman, D.A. Stress-induced premature senescence (SIPS)—Influence of SIPS on radiotherapy. J. Radiat. Res. 2008, 49, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R. Emerging role of radiation induced bystander effects: Cell communications and carcinogenesis. Genome Integr. 2010, 1, 13. [Google Scholar] [CrossRef] [PubMed]
- Blyth, B.J.; Sykes, P.J. Radiation-induced bystander effects: What are they, and how relevant are they to human radiation exposures? Radiat. Res. 2011, 176, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Panganiban, R.A.; Snow, A.L.; Day, R.M. Mechanisms of radiation toxicity in transformed and non-transformed cells. Int. J. Mol. Sci. 2013, 14, 15931–15958. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Fardid, R.; Hadadi, G.; Fardid, M. The mechanisms of radiation-induced bystander effect. J. Biomed. Phys. Eng. 2014, 4, 163–172. [Google Scholar] [PubMed]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kumazaki, T.; Robetorye, R.S.; Robetorye, S.C.; Smith, J.R. Fibronectin expression increases during in vitro cellular senescence: Correlation with increased cell area. Exp. Cell Res. 1991, 195, 13–19. [Google Scholar] [CrossRef]
- Sabin, R.J.; Anderson, R.M. Cellular Senescence—Its role in cancer and the response to ionizing radiation. Genome Integr. 2011, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Vens, C.; Dahmen-Mooren, E.; Verwijs-Janssen, M.; Blyweert, W.; Graversen, L.; Bartelink, H.; Begg, A.C. The role of DNA polymerase beta in determining sensitivity to ionizing radiation in human tumor cells. Nucleic Acids Res. 2002, 30, 2995–3004. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- De Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Brookman, K.W.; Jones, N.J.; Allen, S.A.; Carrano, A.V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell. Biol. 1990, 10, 6160–6171. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.K.; Wilson, D.M., 3rd. XRCC1 and DNA polymerase beta interaction contributes to cellular alkylating-agent resistance and single-strand break repair. J. Cell Biochem. 2005, 95, 794–804. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.G.; Cooper, J.P. Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev. 2004, 18, 2249–2254. [Google Scholar] [CrossRef] [PubMed]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008, 7, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O'Neill, P. Biological consequences of radiation-induced DNA damage: relevance to radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.M.; Xue, L.Y.; Friedman, L.R.; Oleinick, N.L. Comparison of DNA–protein cross-links induced by 4'-(9-acridinylamino)-methanesulfon-m-anisidide and by gamma-radiation. Cancer Res. 1989, 49, 910–914. [Google Scholar] [PubMed]
- Ramakrishnan, N.; Chiu, S.M.; Oteinick, N.L. Yield of DNA–protein cross-links in gamma-irradiated Chinese hamster cells. Cancer Res. 1987, 47, 2032–2035. [Google Scholar] [PubMed]
- Frankenberg-Schwager, M. Induction, repair and biological relevance of radiation-induced DNA lesions in eukaryotic cells. Radiat. Environ. Biophys. 1990, 29, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Kelsey, K.T.; Memisoglu, A.; Frenkel, D.; Liber, H.L. Human lymphocytes exposed to low doses of X-rays are less susceptible to radiation-induced mutagenesis. Mutat. Res. 1991, 263, 197–201. [Google Scholar] [CrossRef]
- Feinendegen, L.E.; Bond, V.P.; Sondhaus, C.A.; Muehlensiepen, H. Radiation effects induced by low doses in complex tissue and their relation to cellular adaptive responses. Mutat. Res. 1996, 358, 199–205. [Google Scholar] [CrossRef]
- Guo, G.; Yan-Sanders, Y.; Lyn-Cook, B.D.; Wang, T.; Tamae, D.; Ogi, J.; Khaletskiy, A.; Li, Z.; Weydert, C.; Longmate, J.A.; et al. Manganese superoxide dismutase-mediated gene expression in radiation-induced adaptive responses. Mol. Cell. Biol. 2003, 23, 2362–2378. [Google Scholar] [CrossRef] [PubMed]
- Pandey, B.N.; Gordon, D.M.; de Toledo, S.M.; Pain, D.; Azzam, E.I. Normal human fibroblasts exposed to high- or low-dose ionizing radiation: Differential effects on mitochondrial protein import and membrane potential. Antioxid. Redox Signal. 2006, 8, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Mammalian G1 cyclins. Cell 1993, 73, 1059–1065. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed]
- Diehl, J.A. Cycling to cancer with cyclin D1. Cancer Biol. Ther. 2002, 1, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Shintani, M.; Okazaki, A.; Masuda, T.; Kawada, M.; Ishizuka, M.; Doki, Y.; Weinstein, I.B.; Imoto, M. Overexpression of cyclin DI contributes to malignant properties of esophageal tumor cells by increasing VEGF production and decreasing Fas expression. Anticancer Res. 2002, 22, 639–647. [Google Scholar] [PubMed]
- Thomas, G.R.; Nadiminti, H.; Regalado, J. Molecular predictors of clinical outcome in patients with head and neck squamous cell carcinoma. Int. J. Exp. Pathol. 2005, 86, 347–363. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Gamble, S.C.; Stavropoulou, A.V.; Pomeranz, K.M.; Lam, E.W.; Coombes, R.C.; Vigushin, D.M. The cyclin D1 proto-oncogene is sequestered in the cytoplasm of mammalian cancer cell lines. Mol. Cancer 2006, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.M.; Fan, M.; Nantajit, D.; Cao, N.; Li, J.J. Cyclin D1 in low-dose radiation-induced adaptive resistance. Oncogene 2008, 27, 6738–6748. [Google Scholar] [CrossRef] [PubMed]
- Camp, E.R.; Li, J.; Minnich, D.J.; Brank, A.; Moldawer, L.L.; MacKay, S.L.; Hochwald, S.N. Inducible nuclear factor-kappaB activation contributes to chemotherapy resistance in gastric cancer. J. Am. Coll. Surg. 2004, 199, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Scartozzi, M.; Bearzi, I.; Pierantoni, C.; Mandolesi, A.; Loupakis, F.; Zaniboni, A.; Catalano, V.; Quadri, A.; Zorzi, F.; Berardi, R.; et al. Nuclear factor-kB tumor expression predicts response and survival in irinotecan-refractory metastatic colorectal cancer treated with cetuximab-irinotecan therapy. J. Clin. Oncol. 2007, 25, 3930–3935. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xia, L.; Lee, L.M.; Khaletskiy, A.; Wang, J.; Wong, J.Y.; Li, J.J. Effector genes altered in MCF-7 human breast cancer cells after exposure to fractionated ionizing radiation. Radiat. Res. 2001, 155, 543–553. [Google Scholar] [CrossRef]
- Ozeki, M.; Tamae, D.; Hou, D.X.; Wang, T.; Lebon, T.; Spitz, D.R.; Li, J.J. Response of cyclin B1 to ionizing radiation: Regulation by NF-kappaB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res. 2004, 24, 2657–2663. [Google Scholar] [PubMed]
- Holley, A.K.; Xu, Y.; St Clair, D.K.; St Clair, W.H. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Ann. N. Y. Acad. Sci. 2010, 1201, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Grdina, D.J.; Murley, J.S.; Miller, R.C.; Mauceri, H.J.; Sutton, H.G.; Li, J.J.; Woloschak, G.E.; Weichselbaum, R.R. A survivin-associated adaptive response in radiation therapy. Cancer Res. 2013, 73, 4418–4428. [Google Scholar] [CrossRef] [PubMed]
- Grdina, D.J.; Murley, J.S.; Miller, R.C.; Woloschak, G.E.; Li, J.J. NFkappaB and Survivin-Mediated Radio-Adaptive Response. Radiat. Res. 2015, 183, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Li, S.; Wang, Z.; Ahmed, K.M.; Degnan, M.E.; Fan, M.; Dynlacht, J.R.; Li, J.J. NF-kappaB-mediated HER2 overexpression in radiation-adaptive resistance. Radiat. Res. 2009, 171, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Chang, T.S.; Jeong, W.; Kim, K. Peroxiredoxin, a novel family of peroxidases. IUBMB Life 2001, 52, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Smith-Pearson, P.S.; Kooshki, M.; Spitz, D.R.; Poole, L.B.; Zhao, W.; Robbins, M.E. Decreasing peroxiredoxin II expression decreases glutathione, alters cell cycle distribution, and sensitizes glioma cells to ionizing radiation and H2O2. Free Radic. Biol. Med. 2008, 45, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tamae, D.; LeBon, T.; Shively, J.E.; Yen, Y.; Li, J.J. The role of peroxiredoxin II in radiation-resistant MCF-7 breast cancer cells. Cancer Res. 2005, 65, 10338–10346. [Google Scholar] [CrossRef] [PubMed]
- Kurimasa, A.; Kumano, S.; Boubnov, N.V.; Story, M.D.; Tung, C.S.; Peterson, S.R.; Chen, D.J. Requirement for the kinase activity of human DNA-dependent protein kinase catalytic subunit in DNA strand break rejoining. Mol. Cell. Biol. 1999, 19, 3877–3884. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, L.; Liu, Y.; Sun, C.; Zhang, H.; Miao, G.; Di, C.X.; Zhou, X.; Zhou, R.; Wang, Z. DNA-PKcs deficiency inhibits glioblastoma cell-derived angiogenesis after ionizing radiation. J. Cell. Physiol. 2015, 230, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.C.; Jackson, S.P. The DNA-dependent protein kinase. Genes Dev. 1999, 13, 916–934. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, A.; Ogawa, H.; Ogawa, T. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 1992, 69, 457–470. [Google Scholar] [CrossRef]
- Daboussi, F.; Dumay, A.; Delacote, F.; Lopez, B.S. DNA double-strand break repair signalling: The case of RAD51 post-translational regulation. Cell Signal. 2002, 14, 969–975. [Google Scholar] [CrossRef]
- Klein, H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008, 7, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Althaus, F.R.; Richter, C. ADP-ribosylation of proteins. Enzymology and biological significance. Mol. Biol. Biochem. Biophys. 1987, 37, 1–237. [Google Scholar] [PubMed]
- Lindahl, T.; Satoh, M.S.; Poirier, G.G.; Klungland, A. Posttranslational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995, 20, 405–411. [Google Scholar] [CrossRef]
- Bernardi, R.; Rossi, L.; Poirier, G.G.; Scovassi, A.I. Analysis of poly(ADP-ribose) glycohydrolase activity in nuclear extracts from mammalian cells. Biochim. Biophys. Acta 1997, 1338, 60–68. [Google Scholar] [CrossRef]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Galande, S.; Kohwi-Shigematso, T. Poly(ADP-ribose) polymerase and Ku autoantigen form a complex and synergistically bind to matrix attachment sequences. J. Biol. Chem. 1999, 274, 20521–20528. [Google Scholar] [CrossRef] [PubMed]
- Ariumi, Y.; Masutani, M.; Copeland, T.D.; Mimori, T.; Sugimura, T.; Shimotohno, K.; Ueda, K.; Hatanaka, M.; Noda, M. Suppression of the poly(ADP-ribose) polymerase activity by DNA-dependent protein kinase in vitro. Oncogene 1999, 18, 4616–4625. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.M.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.Z.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [PubMed]
- Zaremba, T.; Thomas, H.D.; Cole, M.; Coulthard, S.A.; Plummer, E.R.; Curtin, N.J. Poly(ADP-ribose) polymerase-1 (PARP-1) pharmacogenetics, activity and expression analysis in cancer patients and healthy volunteers. Biochem. J. 2011, 436, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Scully, R.; Chen, J.J.; Ochs, R.L.; Keegan, K.; Hoekstra, M.; Feunteun, J.; Livingston, D.M. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell 1997, 90, 425–435. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.M.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA binding by Brca1. Proc. Natl. Acad. Sci. USA 2001, 98, 6086–6091. [Google Scholar] [CrossRef] [PubMed]
- Foray, N.; Marot, D.; Gabriel, A.; Randrianarison, V.; Carr, A.M.; Perricaudet, M.; Ashworth, A.; Jeggo, P. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 2003, 22, 2860–2871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Fan, Q.; Ren, K.Q.; Andreassen, P.R. PALB2 Functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol. Cancer Res. 2009, 7, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ma, J.L.; Wu, J.X.; Ye, L.; Cai, H.; Xia, B.; Yu, X.C. PALB2 Links BRCA1 and BRCA2 in the DNA-damage response. Curr. Biol. 2009, 19, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Front. Oncol. 2013, 3, 113. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, T.J.; Wang, T.; Goel, H.L.; Huang, J.; Stein, G.; Lian, J.; Davis, R.J.; Doxsey, S.; Balaji, K.C.; Aronowitz, J.; et al. Prostate carcinoma and radiation therapy: Therapeutic treatment resistance and strategies for targeted therapeutic intervention. Expert Rev. Anticancer Ther. 2008, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Languino, L.R.; Lian, J.; Stein, G.; Blute, M.; Fitzgerald, T.J. Molecular targets for radiation oncology in prostate cancer. Front. Oncol. 2011, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Fornaro, M.; Moro, L.; Teider, N.; Rhim, J.S.; King, M.; Languino, L.R. Selective modulation of type 1 insulin-like growth factor receptor signaling and functions by beta1 integrins. J. Cell Biol. 2004, 166, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Breen, M.; Zhang, J.; Das, I.; Aznavoorian-Cheshire, S.; Greenberg, N.M.; Elgavish, A.; Languino, L.R. beta1A integrin expression is required for type 1 insulin-like growth factor receptor mitogenic and transforming activities and localization to focal contacts. Cancer Res. 2005, 65, 6692–6700. [Google Scholar] [CrossRef] [PubMed]
- Sayeed, A.; Alam, N.; Trerotola, M.; Languino, L.R. Insulin-like growth factor 1 stimulation of androgen receptor activity requires beta(1A) integrins. J. Cell. Physiol. 2012, 227, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Sayeed, A.; Breen, M.; Zarif, M.J.; Garlick, D.S.; Leav, I.; Davis, R.J.; Fitzgerald, T.J.; Morrione, A.; Hsieh, C.C.; et al. beta1 integrins mediate resistance to ionizing radiation in vivo by inhibiting c-Jun amino terminal kinase 1. J. Cell. Physiol. 2013, 228, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Eke, I.; Deuse, Y.; Hehlgans, S.; Gurtner, K.; Krause, M.; Baumann, M.; Shevchenko, A.; Sandfort, V.; Cordes, N. beta(1)Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Investig. 2012, 122, 1529–1540. [Google Scholar] [CrossRef] [PubMed]
- Pawar, S.C.; Dougherty, S.; Pennington, M.E.; Demetriou, M.C.; Stea, B.D.; Dorr, R.T.; Cress, A.E. alpha6 integrin cleavage: Sensitizing human prostate cancer to ionizing radiation. Int. J. Radiat. Biol. 2007, 83, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Han, E.K.; McGonigal, T. Role of focal adhesion kinase in human cancer: A potential target for drug discovery. Anticancer Agents Med. Chem. 2007, 7, 681–684. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, T.; Koguchi, E.; Funakoshi, M.; Aizu-Yokota, E.; Sonoda, Y. Antiapoptotic action of focal adhesion kinase (FAK) against ionizing radiation. Antioxid. Redox Signal. 2002, 4, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.E.; Glenney, J.R., Jr.; Burridge, K. Paxillin: A new vinculin-binding protein present in focal adhesions. J. Cell Biol. 1990, 111, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Bellis, S.L.; Miller, J.T.; Turner, C.E. Characterization of tyrosine phosphorylation of paxillin in vitro by focal adhesion kinase. J. Biol. Chem. 1995, 270, 17437–17441. [Google Scholar] [CrossRef] [PubMed]
- Seidler, J.; Durzok, R.; Brakebusch, C.; Cordes, N. Interactions of the integrin subunit beta1A with protein kinase B/Akt, p130Cas and paxillin contribute to regulation of radiation survival. Radiother. Oncol. 2005, 76, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.T.; Chen, M.F.; Chen, W.C.; Hsieh, C.C. The role of IL-6 in the radiation response of prostate cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Di Maggio, F.M.; Minafra, L.; Forte, G.I.; Cammarata, F.P.; Lio, D.; Messa, C.; Gilardi, M.C.; Bravata, V. Portrait of inflammatory response to ionizing radiation treatment. J. Inflamma. Lond. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Pasi, F.; Facoetti, A.; Nano, R. IL-8 and IL-6 bystander signalling in human glioblastoma cells exposed to gamma radiation. Anticancer Res. 2010, 30, 2769–2772. [Google Scholar] [PubMed]
- Biswas, S.; Guix, M.; Rinehart, C.; Dugger, T.C.; Chytil, A.; Moses, H.L.; Freeman, M.L.; Arteaga, C.L. Inhibition of TGF-beta with neutralizing antibodies prevents radiation-induced acceleration of metastatic cancer progression. J. Clin. Investig. 2007, 117, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.; Iyengar, P.; Pandita, T.K. The role of inflammatory pathways in cancer-associated cachexia and radiation resistance. Mol. Cancer Res. 2013, 11, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Mutter, R.W.; Cao, C.; Albert, J.M.; Shinohara, E.T.; Sekhar, K.R.; Lu, B. Inhibition of signal transducer and activator of transcription 3 activity results in down-regulation of Survivin following irradiation. Mol. Cancer Ther. 2006, 5, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, I.; Skvortsov, S.; Stasyk, T.; Raju, U.; Popper, B.A.; Schiestl, B.; von Guggenberg, E.; Neher, A.; Bonn, G.K.; Huber, L.A.; et al. Intracellular signaling pathways regulating radioresistance of human prostate carcinoma cells. Proteomics 2008, 8, 4521–4533. [Google Scholar] [CrossRef] [PubMed]
- Belenkov, A.I.; Shenouda, G.; Rizhevskaya, E.; Cournoyer, D.; Belzile, J.P.; Souhami, L.; Devic, S.; Chow, T.Y.K. Erythropoietin induces cancer cell resistance to ionizing radiation and to cisplatin. Mol. Cancer Ther. 2004, 3, 1525–1532. [Google Scholar] [PubMed]
- Reader, J.; Holt, D.; Fulton, A. Prostaglandin E-2 EP receptors as therapeutic targets in breast cancer. Cancer Metastasis Rev. 2011, 30, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Zhao, Y.; Yu, S.Y.; Zhang, M.X. Activated PI3K/Akt/COX-2 pathway induces resistance to radiation in human cervical cancer hela cells. Cancer Biother. Radiopharm. 2010, 25, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [PubMed]
- Varjosalo, M.; Taipale, J. Hedgehog: Functions and mechanisms. Genes Dev. 2008, 22, 2454–2472. [Google Scholar] [CrossRef] [PubMed]
- Medina, V.; Calvo, M.B.; Diaz-Prado, S.; Espada, J. Hedgehog signalling as a target in cancer stem cells. Clin. Transl. Oncol. 2009, 11, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Gulino, A.; Ferretti, E.; De Smaele, E. Hedgehog signalling in colon cancer and stem cells. EMBO Mol. Med. 2009, 1, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Altaba, A.R.I. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.F.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Kubo, M.; Nakamura, M.; Tasaki, A.; Yamanaka, N.; Nakashima, H.; Nomura, M.; Kuroki, S.; Katano, M. Hedgehog signaling pathway is a new therapeutic target for patients with breast cancer. Cancer Res. 2004, 64, 6071–6074. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.L.; Chen, K.; Huang, S.H.; Zhang, X.L.; Adegboyega, P.A.; Evers, B.M.; Zhang, H.W.; Xie, J.W. Frequent activation of the hedgehog pathway in advanced gastric adenocarcinomas. Carcinogenesis 2005, 26, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (Review). Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.J.; Ma, J.G.; Ma, Q.Y.; Li, X.Q.; Liu, H.; Xu, Q.H.; Duan, W.X.; Sun, Q.; Xu, J.; Wu, Z.; et al. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Eagles, J.; Keysar, S.B.; Wang, G.L.; Glogowska, M.J.; Altunbas, C.; Anderson, R.T.; Le, P.N.; Morton, J.J.; Frederick, B.; et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014, 74, 7024–7036. [Google Scholar] [CrossRef] [PubMed]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar] [PubMed]
- Qian, L.W.; Mizumoto, K.; Urashima, T.; Nagai, E.; Maehara, N.; Sato, N.; Nakajima, M.; Tanaka, M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 2002, 8, 1223–1227. [Google Scholar] [PubMed]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Nager, M.; Bhardwaj, D.; Canti, C.; Medina, L.; Nogues, P.; Herreros, J. beta-Catenin Signalling in Glioblastoma Multiforme and Glioma-Initiating Cells. Chemother. Res. Pract. 2012, 2012, 192362. [Google Scholar] [PubMed]
- Kim, Y.; Kim, K.H.; Lee, J.; Lee, Y.A.; Kim, M.; Lee, S.J.; Park, K.; Yang, H.; Jin, J.; Joo, K.M.; et al. Wnt activation is implicated in glioblastoma radioresistance. Lab. Investig. 2012, 92, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed]
- VanDussen, K.L.; Carulli, A.J.; Keeley, T.M.; Patel, S.R.; Puthoff, B.J.; Magness, S.T.; Tran, I.T.; Maillard, I.; Siebel, C.; Kolterud, A.; et al. Notch signaling modulates proliferation and differentiation of intestinal crypt base columnar stem cells. Development 2012, 139, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Raj, K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat. Rev. Cancer 2003, 3, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Oh, P.; Aifantis, I. Oncogenic and tumor suppressor functions of Notch in cancer: It's NOTCH what you think. J. Exp. Med. 2011, 208, 1931–1935. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.L.; Kissil, J.L. Notch signaling in pancreatic cancer: Oncogene or tumor suppressor? Trends Mol. Med. 2013, 19, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Capaccione, K.M.; Pine, S.R. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis 2013, 34, 1420–1430. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The response of CD24(−/low)/CD44(+) breast cancer-initiating cells to radiation. J. Natl. Cancer Inst. 2006, 98, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Theys, J.; Yahyanejad, S.; Habets, R.; Span, P.; Dubois, L.; Paesmans, K.; Kattenbeld, B.; Cleutjens, J.; Groot, A.J.; Schuurbiers, O.C.J.; et al. High NOTCH activity induces radiation resistance in non small cell lung cancer. Radiother. Oncol. 2013, 108, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Nordsmark, M.; Overgaard, M.; Overgaard, J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiother. Oncol. 1996, 41, 31–39. [Google Scholar] [CrossRef]
- Brizel, D.M.; Sibley, G.S.; Prosnitz, L.R.; Scher, R.L.; Dewhirst, M.W. Tumor hypoxia adversely affects the prognosis of carcinoma of the head and neck. Int. J. Radiat. Oncol. Biol. Phys. 1997, 38, 285–289. [Google Scholar] [CrossRef]
- Harada, H.; Kizaka-Kondoh, S.; Li, G.; Itasaka, S.; Shibuya, K.; Inoue, M.; Hiraoka, M. Significance of HIF-1-active cells in angiogenesis and radioresistance. Oncogene 2007, 26, 7508–7516. [Google Scholar] [CrossRef] [PubMed]
- Kessler, J.; Hahnel, A.; Wichmann, H.; Rot, S.; Kappler, M.; Bache, M.; Vordermark, D. HIF-1 alpha inhibition by siRNA or chetomin in human malignant glioma cells: Effects on hypoxic radioresistance and monitoring via CA9 expression. BMC Cancer 2010, 10. [Google Scholar] [CrossRef] [PubMed]
- Harada, H. How Can We Overcome Tumor Hypoxia in Radiation Therapy? J. Radiat. Res. 2011, 52, 545–556. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1 alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Neufeld, G.; Cohen, T.; Gengrinovitch, S.; Poltorak, Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999, 13, 9–22. [Google Scholar] [PubMed]
- McMahon, G. VEGF receptor signaling in tumor angiogenesis. Oncologist 2000, 5, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Noordhuis, M.G.; Eijsink, J.J.H.; ten Hoor, K.A.; Roossink, F.; Hollema, H.; Arts, H.J.G.; Pras, E.; Maduro, J.H.; Reyners, A.K.L.; de Bock, G.H.; et al. Expression of epidermal growth factor receptor (EGFR) and activated EGFR predict poor response to (chemo)radiation and survival in cervical cancer. Clin. Cancer Res. 2009, 15, 7389–7397. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Okamoto, I.; Kimura, H.; Sakai, K.; Nishimura, Y.; Nishio, K.; Nakagawa, K. Clinical outcomes of thoracic radiotherapy for locally advanced NSCLC with EGFR mutations or EML4-ALK rearrangement. Anticancer Res. 2012, 32, 4533–4537. [Google Scholar] [PubMed]
- Rodemann, H.P.; Blaese, M.A. Responses of normal cells to ionizing radiation. Semin. Radiat. Oncol. 2007, 17, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Rodemann, H.P.; Dittmann, K.; Toulany, M. Radiation-induced EGFR-signaling and control of DNA-damage repair. Int. J. Radiat. Biol. 2007, 83, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Rodemann, H.P. Membrane receptor signaling and control of DNA repair after exposure to ionizing radiation. Nuklearmedizin 2010, 49, S26–S30. [Google Scholar] [PubMed]
- Valerie, K.; Yacoub, A.; Hagan, M.P.; Curiel, D.T.; Fisher, P.B.; Grant, S.; Dent, P. Radiation-induced cell signaling: Inside-out and outside-in. Mol. Cancer Ther. 2007, 6, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Li, H.F.; Kim, J.S.; Waldman, T. Radiation-induced Akt activation modulates radioresistance in human glioblastoma cells. Radiat. Oncol. 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Minjgee, M.; Kehlbach, R.; Chen, J.Y.; Baumann, M.; Rodemann, H.P. ErbB2 expression through heterodimerization with erbB1 is necessary for ionizing radiation—But not EGF-induced activation of Akt survival pathway. Radiother. Oncol. 2010, 97, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Brognard, J.; Clark, A.S.; Ni, Y.C.; Dennis, P.A. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 2001, 61, 3986–3997. [Google Scholar] [PubMed]
- Noguchi, M.; Hirata, N.; Suizu, F. The links between AKT and two intracellular proteolytic cascades: Ubiquitination and autophagy. Biochim. Biophys. Acta 2014, 1846, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Wilson, E.N.; Bristol, M.L.; Di, X.; Maltese, W.A.; Koterba, K.; Beckman, M.J.; Gewirtz, D.A. A switch between cytoprotective and cytotoxic autophagy in the radiosensitization of breast tumor cells by chloroquine and vitamin D. Horm. Cancer 2011, 2, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin D-3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, K.; Iwado, E.; Mills, G.B.; Sawaya, R.; Kondo, S.; Kondo, Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int. J. Oncol. 2007, 31, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Lee, K.J.; Fattah, K.R.; Lin, Y.F.; Fehrenbacher, B.; Schaller, M.; Chen, B.P.; Chen, D.J.; Rodemann, H.P. Akt Promotes Post-irradiation survival of human tumor cells through initiation, progression, and termination of DNA-PKcs-dependent DNA double-strand break repair. Mol. Cancer Res. 2012, 10, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Bozulic, L.; Surucu, B.; Hynx, D.; Hemmings, B.A. PKB alpha/Akt1 acts downstream of DNA-PK in the DNA double-strand break response and promotes survival. Mol. Cell 2008, 30, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Harding, S.M.; Zhao, H.; Coackley, C.; Durocher, D.; Bristow, R.G. MRE11 promotes AKT phosphorylation in direct response to DNA double-strand breaks. Cell Cycle 2011, 10, 2218–2232. [Google Scholar] [CrossRef] [PubMed]
- Plo, I.; Laulier, C.; Gauthier, L.; Lebrun, F.; Calvo, F.; Lopez, B.S. AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 2008, 68, 9404–9412. [Google Scholar] [CrossRef] [PubMed]
- Albert, J.M.; Kim, K.W.; Cao, C.; Lu, B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther. 2006, 5, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis. 2014, 5, e1437. [Google Scholar] [CrossRef] [PubMed]
- Kao, G.D.; Jiang, Z.; Fernandes, A.M.; Gupta, A.K.; Maity, A. Inhibition of phosphatidylinositol-3-OH kinase/Akt signaling impairs DNA repair in glioblastoma cells following ionizing radiation. J. Biol. Chem. 2007, 282, 21206–21212. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Q.; Jiang, Y.; Li, G. Inhibition of the PI3K/AKT-NF-kappaB pathway with curcumin enhanced radiation-induced apoptosis in human Burkitt's lymphoma. J. Pharmacol. Sci. 2013, 121, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Chautard, E.; Loubeau, G.; Tchirkov, A.; Chassagne, J.; Vermot-Desroches, C.; Morel, L.; Verrelle, P. Akt signaling pathway: A target for radiosensitizing human malignant glioma. Neuro Oncol. 2010, 12, 434–443. [Google Scholar] [PubMed]
- Liu, W.L.; Gao, M.; Tzen, K.Y.; Tsai, C.L.; Hsu, F.M.; Cheng, A.L.; Cheng, J.C.H. Targeting Phosphatidylinositide3-Kinase/Akt pathway by BKM120 for radiosensitization in hepatocellular carcinoma. Oncotarget 2014, 5, 3662–3672. [Google Scholar] [CrossRef] [PubMed]
- Ekshyyan, O.; Rong, Y.H.; Rong, X.H.; Pattani, K.M.; Abreo, F.; Caldito, G.; Chang, J.K.S.; Arnpil, F.; Glass, J.; Nathan, C.A.O. Comparison of radiosensitizing effects of the mammalian target of rapamycin inhibitor CCI-779 to cisplatin in experimental models of head and neck squamous cell carcinoma. Mol. Cancer Ther. 2009, 8, 2255–2265. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Myers, C.J.; Jung, D.K.; Lu, B. NVP-BEZ-235 enhances radiosensitization via blockade of the PI3K/mTOR pathway in cisplatin-resistant non-small cell lung carcinoma. Genes Cancer 2014, 5, 293–302. [Google Scholar] [PubMed]
- Ma, Z.K.; Ya, G.L.; Zhou, B.; Fan, Y.G.; Gao, S.G.; Feng, X.S. The Chk1 inhibitor AZD7762 sensitises p53 mutant breast cancer cells to radiation in vitro and in vivo. Mol. Med. Rep. 2012, 6, 897–903. [Google Scholar] [PubMed]
- Borst, G.R.; McLaughlin, M.; Kyula, J.N.; Neijenhuis, S.; Khan, A.; Good, J.; Zaidi, S.; Powell, N.G.; Meier, P.; Collins, I.; et al. Targeted radiosensitization by the Chk1 inhibitor SAR-020106. Int. J. Radiat. Oncol. Biol. Phys. 2013, 85, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; Parsels, L.A.; Zhao, L.L.; Parsels, J.D.; Davis, M.A.; Hassan, M.C.; Arumugarajah, S.; Hylander-Gans, L.; Morosini, D.; Simeone, D.M.; et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G(2) checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010, 70, 4972–4981. [Google Scholar] [CrossRef] [PubMed]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Chiao, P.J.; Andreeff, M.; Meyn, R.E. Inhibition of constitutively activated nuclear factor-kappa B radiosensitizes human melanoma cells. Mol. Cancer Ther. 2004, 3, 985–992. [Google Scholar] [PubMed]
- Kim, B.Y.; Kim, K.A.; Kwon, O.; Kim, S.O.; Kim, M.S.; Kim, B.S.; Oh, W.K.; Kim, G.D.; Jung, M.; Ahn, J.S. NF-kappa B inhibition radiosensitizes Ki-Ras-transformed cells to ionizing radiation. Carcinogenesis 2005, 26, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, Y.; Kurimoto, M.; Nagai, S.; Hayakawa, Y.; Kamiyama, H.; Hayashi, N.; Kitajima, I.; Endo, S. Induction of autophagic cell death and radiosensitization by the pharmacological inhibition of nuclear factor-kappa B activation in human glioma cell lines. J. Neurosurg. 2009, 110, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.R.; Efimova, E.V.; Hamzeh, K.W.; Darga, T.E.; Mauceri, H.J.; Fu, Y.X.; Kron, S.J.; Weichselbaum, R.R. Radiation-inducible immunotherapy for cancer: senescent tumor cells as a cancer vaccine. Mol. Ther. 2012, 20, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Wilken, R.; Veena, M.S.; Wang, M.B.; Srivatsan, E.S. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol. Cancer 2011, 10. [Google Scholar] [CrossRef]
- Glienke, W.; Maute, L.; Wicht, J.; Bergmann, L. Curcumin inhibits constitutive STAT3 phosphorylation in human pancreatic cancer cell lines and downregulation of survivin/BIRC5 gene expression. Cancer Investig. 2010, 28, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Chendil, D.; Ranga, R.S.; Meigooni, D.; Sathishkumar, S.; Ahmed, M.M. Curcumin confers radiosensitizing effect in prostate cancer cell line PC-3. Oncogene 2004, 23, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Garg, A.K.; Buchholz, T.A.; Aggarwal, B.B. Chemosensitization and radiosensitization of tumors by plant polyphenols. Antioxid. Redox Signal. 2005, 7, 1630–1647. [Google Scholar] [CrossRef] [PubMed]
- Okunieff, P.; Xu, J.H.; Hu, D.P.; Liu, W.M.; Zhang, L.R.; Morrow, G.; Pentland, A.; Ryan, J.L.; Ding, I. Curcumin protects against radiation-induced acute and chronic cutaneous toxicity in mice and decreases mRNA expression of inflammatory and fibrogenic cytokines. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Seong, K.M.; Youn, B. Phenylpropanoids in radioregulation: Double edged sword. Exp. Mol. Med. 2011, 43, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.; Liu, C.Q.; Sanli, T.; Tsiani, E.; Singh, G.; Bristow, R.G.; Dayes, I.; Lukka, H.; Wright, J.; Tsakiridis, T. Resveratrol enhances prostate cancer cell response to ionizing radiation. Modulation of the AMPK, Akt and mTOR pathways. Radiat. Oncol. 2011, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Kma, L. Synergistic effect of resveratrol and radiotherapy in control of cancers. Asian Pac. J. Cancer Prev. 2013, 14, 6197–6208. [Google Scholar] [CrossRef] [PubMed]
- Scarlatti, F.; Sala, G.; Ricci, C.; Maioli, C.; Milani, F.; Minella, M.; Botturi, M.; Ghidoni, R. Resveratrol sensitization of DU145 prostate cancer cells to ionizing radiation is associated to ceramide increase. Cancer Lett. 2007, 253, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.M.; Wang, L.; Schulte, B.A.; Yang, A.M.; Tang, S.S.; Wang, G.Y. Resveratrol enhances ionizing radiation-induced premature senescence in lung cancer cells. Int. J. Oncol. 2013, 43, 1999–2006. [Google Scholar] [PubMed]
- Carsten, R.E.; Bachand, A.M.; Bailey, S.M.; Ullrich, R.L. Resveratrol reduces radiation-induced chromosome aberration frequencies in mouse bone marrow cells. Radiat. Res. 2008, 169, 633–638. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, T.; Nonaka, T.; Ishikawa, H.; Sakurai, H.; Saitoh, J.; Takahashi, T.; Mitsuhashi, N. Genistein, a tyrosine kinase inhibitor, enhanced radiosensitivity in human esophageal cancer cell lines in vitro: Possible involvement of inhibition of survival signal transduction pathways. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 195–201. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.Y.; Pan, J.S.; Han, S.P.; Yin, X.X.; Wang, B.; Hu, G. Combined treatment of ionizing radiation with genistein on cervical cancer HeLa cells. J. Pharmacol. Sci. 2006, 102, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Jeong, M.H.; Jin, Y.H.; Kang, E.Y.; Jo, W.S.; Park, H.T.; Lee, J.D.; Yoo, Y.J.; Jeong, S.J. The modulation of radiation-induced cell death by genistein in K562 cells: Activation of thymidine kinase 1. Cell Res. 2004, 14, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Raffoul, J.J.; Wang, Y.; Kucuk, O.; Forman, J.D.; Sarkar, F.H.; Hillman, G.G. Genistein inhibits radiation-induced activation of NF-kappa B in prostate cancer cells promoting apoptosis and G(2)/M cell cycle arrest. BMC Cancer 2006, 6, 107. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.G.; Kim, J.S.; Lee, J.H.; Cho, E.W. Genistein decreases cellular redox potential, partially suppresses cell growth in HL-60 leukemia cells and sensitizes cells to gamma-radiation-induced cell death. Mol. Med. Rep. 2014, 10, 2786–2792. [Google Scholar] [PubMed]
- Priyadarsini, R.V.; Murugan, R.S.; Maitreyi, S.; Ramalingam, K.; Karunagaran, D.; Nagini, S. The flavonoid quercetin induces cell cycle arrest and mitochondria-mediated apoptosis in human cervical cancer (HeLa) cells through p53 induction and NF-kappa B inhibition. Eur. J. Pharmacol. 2010, 649, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Moon, J.Y.; Ahn, K.S.; Cho, S.K. Quercetin induces mitochondrial mediated apoptosis and protective autophagy in human glioblastoma U373MG cells. Oxidative Med. Cell. Longev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Sudan, S.; Rupasinghe, H.P.V. Quercetin-3-O-glucoside Induces Human DNA Topoisomerase II Inhibition, cell cycle arrest and apoptosis in hepatocellular carcinoma cells. Anticancer Res. 2014, 34, 1691–1699. [Google Scholar] [PubMed]
- Lin, C.H.; Yu, Y.; Zhao, H.G.; Yang, A.M.; Yan, H.; Cui, Y.L. Combination of quercetin with radiotherapy enhances tumor radiosensitivity in vitro and in vivo. Radiother. Oncol. 2012, 104, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Benkovic, V.; Knezevic, A.H.; Dikic, D.; Lisicic, D.; Orsolic, N.; Basic, I.; Kopjar, N. Radioprotective effects of quercetin and ethanolic extract of propolis in gamma-irradiated mice. Arch. Ind. Hyg. Toxicol. 2009, 60, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xue, J.X.; Li, X.; Ao, R.; Lu, Y. Quercetin liposomes protect against radiation-induced pulmonary injury in a murine model. Oncol. Lett. 2013, 6, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.R.; Beckett, M.A.; Liang, H.; Mauceri, H.J.; van Rooijen, N.; Cohen, K.S.; Weichselbaum, R.R. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010, 70, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining radiotherapy and cancer immunotherapy: A paradigm shift. J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, J. Hypoxic radiosensitization: Adored and ignored. J. Clin. Oncol. 2007, 25, 4066–4074. [Google Scholar] [CrossRef] [PubMed]
- Meijer, T.W.H.; Kaanders, J.H.A.M.; Span, P.N.; Bussink, J. Targeting hypoxia, HIF-1, and tumor glucose metabolism to improve radiotherapy efficacy. Clin. Cancer Res. 2012, 18, 5585–5594. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Shin, D.H.; Chun, Y.S.; Lee, M.K.; Kim, M.S.; Park, J.W. A novel mode of action of YC-1 in HIF inhibition: Stimulation of FIH-dependent p300 dissociation from HIF-1 alpha. Mol. Cancer Ther. 2008, 7, 3729–3738. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.J.; Chun, Y.S.; Cho, Y.S.; Kim, J.H.; Lee, J.C.; Kim, M.S.; Park, J.W. YGI: A potential anticancer drug targeting hypoxia-inducible factor 1. J. Natl. Cancer Inst. 2003, 95, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.H.; Kim, J.H.; Jung, Y.J.; Kim, K.E.; Jeong, J.M.; Chun, Y.S.; Park, J.W. Preclinical evaluation of YG-1, a HIF inhibitor, for the prevention of tumor spreading. Cancer Lett. 2007, 255, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Inoue, M.; Itasaka, S.; Hirota, K.; Morinibu, A.; Shinomiya, K.; Zeng, L.H.; Ou, G.F.; Zhu, Y.X.; Yoshimura, M.; et al. Cancer cells that survive radiation therapy acquire HIF-1 activity and translocate towards tumour blood vessels. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallio, P.J.; Pongratz, I.; Gradin, K.; McGuire, J.; Poellinger, L. Activation of hypoxia-inducible factor 1 alpha: Posttranscriptional regulation and conformational change by recruitment of the Arnt transcription factor. Proc. Natl. Acad. Sci. USA 1997, 94, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Zhang, H.F.; Qian, D.Z.; Rey, S.; Liu, J.O.; Semenza, G.L. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc. Natl. Acad. Sci. USA 2009, 106, 17910–17915. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.V.; Baek, J.H.; Zhang, H.; Diez, R.; Cole, R.N.; Semenza, G.L. RACK1 competes with HSP90 for binding to HIF-1 alpha and is required for O-2-independent and HSP90 inhibitor-induced degradation of HIF-1 alpha. Mol. Cell 2007, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.Y.; Oh, S.H.; Woo, J.K.; Hong, W.K.; Lee, H.Y. Targeting heat shock protein 90 overrides the resistance of lung cancer cells by blocking radiation-induced stabilization of hypoxia-inducible factor-1 alpha. Cancer Res. 2009, 69, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Nieder, C.; Wiedenmann, N.; Andratschke, N.; Molls, M. Current status of angiogenesis inhibitors combined with radiation therapy. Cancer Treat. Rev. 2006, 32, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Dings, R.P.M.; Loren, M.; Heun, H.; McNiel, E.; Griffioen, A.W.; Mayo, K.H.; Griffin, R.J. Scheduling of radiation with angiogenesis inhibitors anginex and avastin improves therapeutic outcome via vessel normalization. Clin. Cancer Res. 2007, 13, 3395–3402. [Google Scholar] [CrossRef] [PubMed]
- Kleibeuker, E.A.; Griffioen, A.W.; Verheul, H.M.; Slotman, B.J.; Thijssen, V.L. Combining angiogenesis inhibition and radiotherapy: A double-edged sword. Drug Resist. Updates 2012, 15, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Choy, H.; Milas, L. Enhancing radiotherapy with cyclooxygenase-2 enzyme inhibitors: A rational advance? J. Natl. Cancer Inst. 2003, 95, 1440–1452. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.M.; Hong, Y.; Lee, S.; Liu, P.; Lim, J.H.; Lee, Y.H.; Lee, T.H.; Chang, K.T.; Hong, Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. Int. J. Mol. Sci. 2015, 16, 26880-26913. https://doi.org/10.3390/ijms161125991
Kim BM, Hong Y, Lee S, Liu P, Lim JH, Lee YH, Lee TH, Chang KT, Hong Y. Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. International Journal of Molecular Sciences. 2015; 16(11):26880-26913. https://doi.org/10.3390/ijms161125991
Chicago/Turabian StyleKim, Byeong Mo, Yunkyung Hong, Seunghoon Lee, Pengda Liu, Ji Hong Lim, Yong Heon Lee, Tae Ho Lee, Kyu Tae Chang, and Yonggeun Hong. 2015. "Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy" International Journal of Molecular Sciences 16, no. 11: 26880-26913. https://doi.org/10.3390/ijms161125991
APA StyleKim, B. M., Hong, Y., Lee, S., Liu, P., Lim, J. H., Lee, Y. H., Lee, T. H., Chang, K. T., & Hong, Y. (2015). Therapeutic Implications for Overcoming Radiation Resistance in Cancer Therapy. International Journal of Molecular Sciences, 16(11), 26880-26913. https://doi.org/10.3390/ijms161125991