
Multi-Functional Macromers for Hydrogel Design in Biomedical Engineering and Regenerative Medicine

Abstract
:1. Introduction
2. Macromers with at Least Two Types of Functional Groups for Cross-Linking
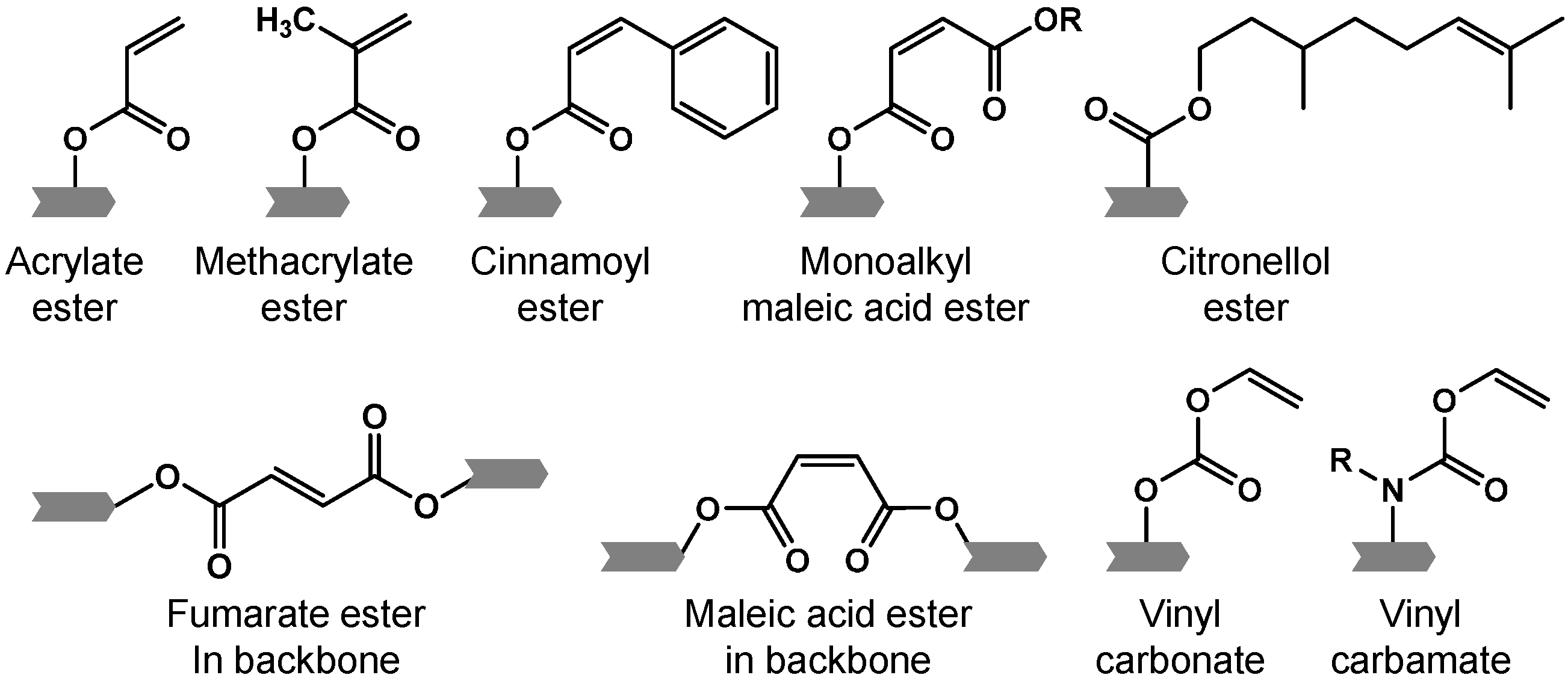
 represents an oligomer segment that contains a functional group.
represents an oligomer segment that contains a functional group.
represents an oligomer segment that contains a functional group.
represents an oligomer segment that contains a functional group.
2.1. Macromers with Dual (Physical and Chemical) Gelation Properties
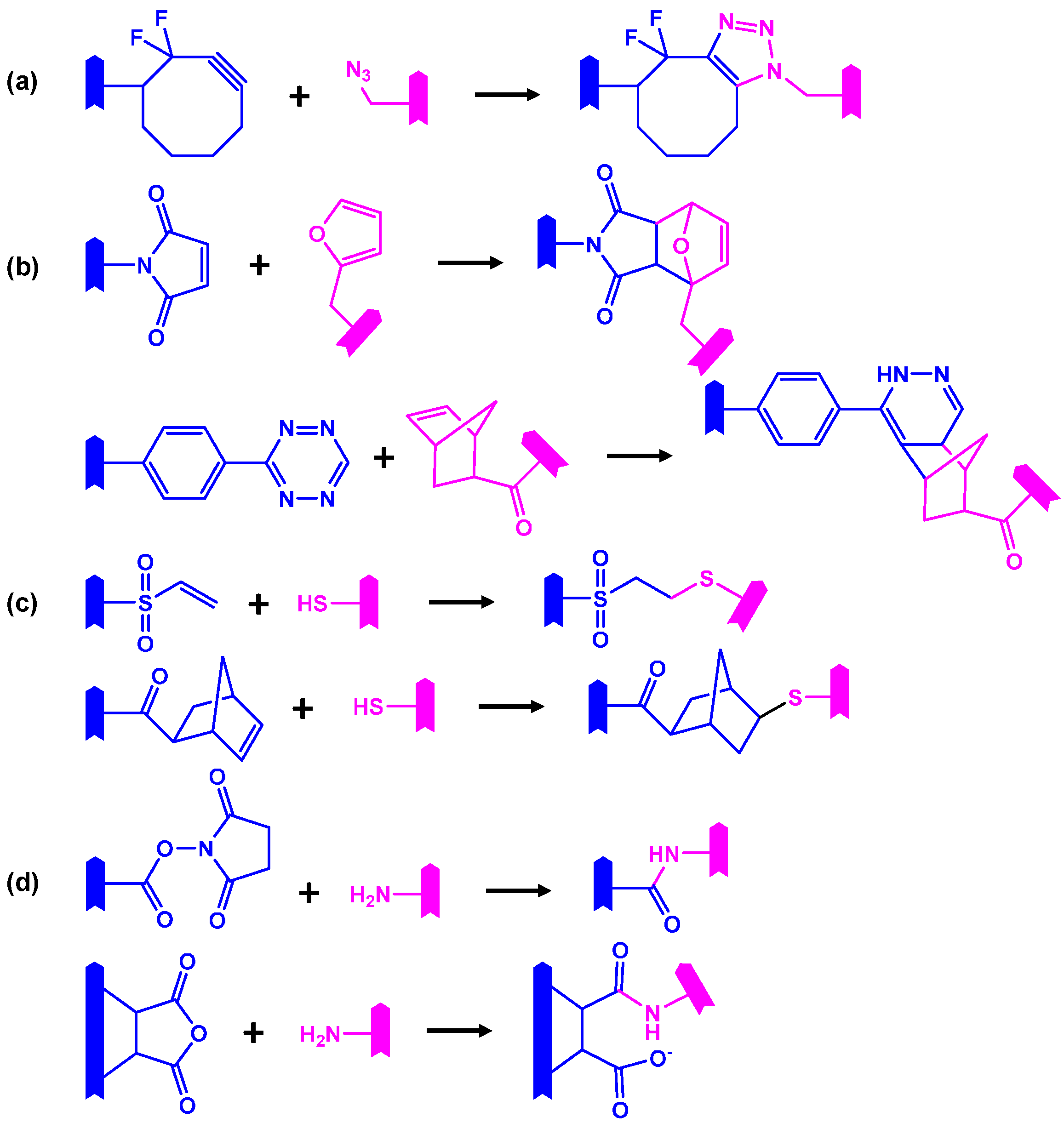
represents an oligomer segment that contains a functional group. (a) Strain-promoted metal-free cycloaddition of a difluorocyclooctyne with an azide; (b) Diels-Alder cycloaddition of a maleimide with a furane and below an Inverse electron demand Diels–Alder reaction of a tetrazine with a norbornene; (c) Thiol-ene reaction of a vinyl sulfone with a thiol through a Michael addition mechanism and below a free-radical thiol-ene addition of a thiol to a norbornene; (d) Amide formation of a primary amine with a succinimide ester or a cyclic anhydride (below).
represents an oligomer segment that contains a functional group. (a) Strain-promoted metal-free cycloaddition of a difluorocyclooctyne with an azide; (b) Diels-Alder cycloaddition of a maleimide with a furane and below an Inverse electron demand Diels–Alder reaction of a tetrazine with a norbornene; (c) Thiol-ene reaction of a vinyl sulfone with a thiol through a Michael addition mechanism and below a free-radical thiol-ene addition of a thiol to a norbornene; (d) Amide formation of a primary amine with a succinimide ester or a cyclic anhydride (below).
2.1.1. NiPAAm-Based Dual-Gelling Macromers

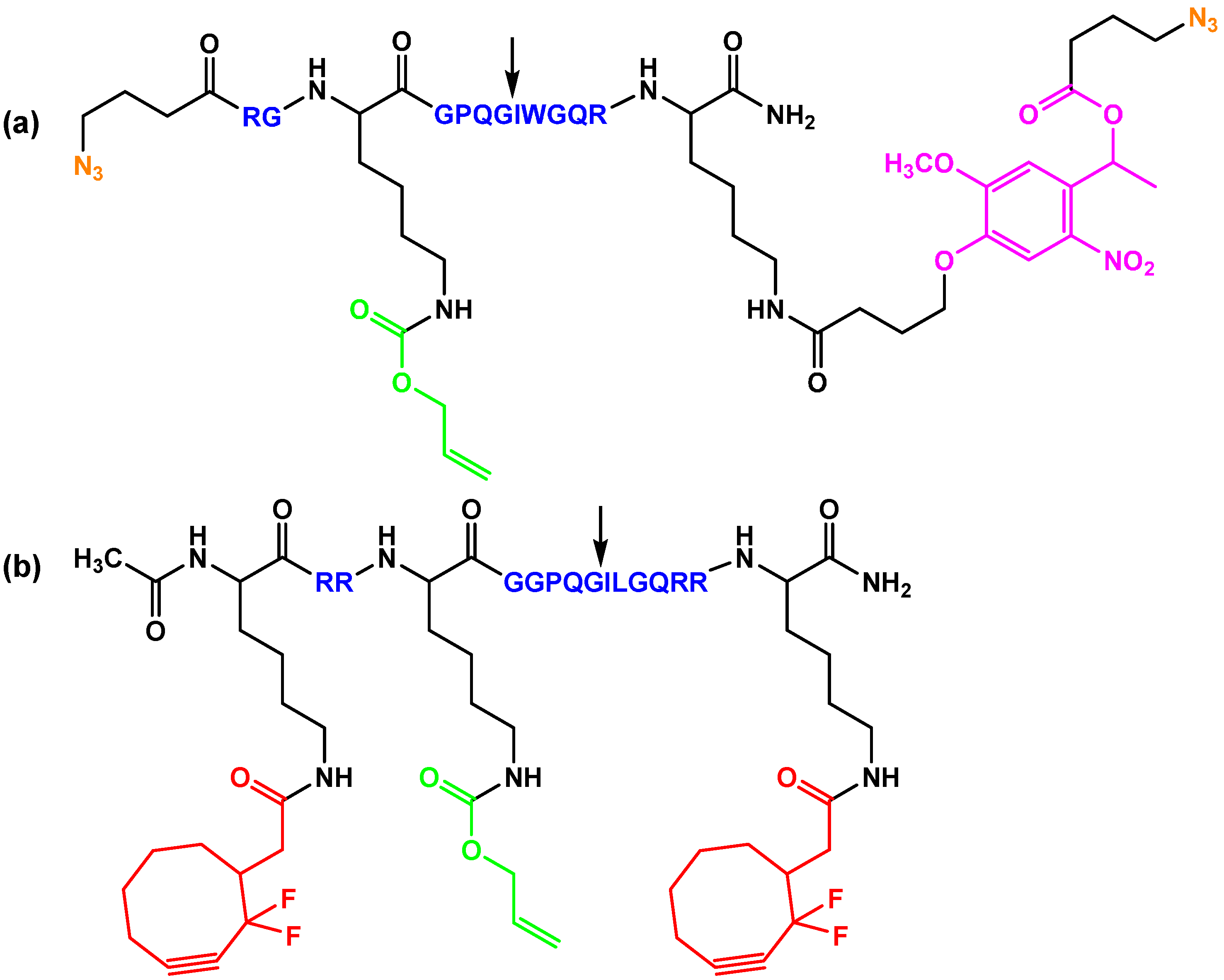
 represents an oligomer segment that contains a functional group (blue, magenta). The initial reaction is a reversible transthioesterification step which is chemoselective and regioselective. The thioester intermediate rearranges by an intramolecular S,N-acyl shift that results in the formation of a stable native amide. Note that the resulting network provides fee thiol moieties for additional functionalization reactions.
represents an oligomer segment that contains a functional group (blue, magenta). The initial reaction is a reversible transthioesterification step which is chemoselective and regioselective. The thioester intermediate rearranges by an intramolecular S,N-acyl shift that results in the formation of a stable native amide. Note that the resulting network provides fee thiol moieties for additional functionalization reactions.
represents an oligomer segment that contains a functional group (blue, magenta). The initial reaction is a reversible transthioesterification step which is chemoselective and regioselective. The thioester intermediate rearranges by an intramolecular S,N-acyl shift that results in the formation of a stable native amide. Note that the resulting network provides fee thiol moieties for additional functionalization reactions.
represents an oligomer segment that contains a functional group (blue, magenta). The initial reaction is a reversible transthioesterification step which is chemoselective and regioselective. The thioester intermediate rearranges by an intramolecular S,N-acyl shift that results in the formation of a stable native amide. Note that the resulting network provides fee thiol moieties for additional functionalization reactions.
2.1.2. Copolymer-Based Dual-Gelling Macromers
2.1.3. Poly(organophosphazene) (POP)-Based Dual-Gelling Macromers
3. Oligo-Functional Macromers with at Least One Type of Functionality not Involved in Cross-Linking
3.1. Thermogelling Macromers with at Least One Type of Functionality not Involved in Cross-Linking
3.2. Chemically Cross-Linkable Macromers with at Least One Type of Functionality not Involved in Cross-Linking
3.2.1. Chemically Cross-Linkable Macromers with More Than One Functionality for Cross-Linking and/or Bioconjugation
3.2.2. Chemically Cross-Linkable Macromers with Cell Adhesive Motifs
3.2.3. Chemically Cross-Linkable Macromers with Other Additional Properties
3.3. Macromers with Advanced Degradative Properties
3.3.1. Oligofunctional Macromers Sensitive to Reductive or Enzymatic Degradation
3.3.2. Photodegradable Oligofunctional Macromers
3.4. Hydrogel-Forming Macromers with Shape Memory Properties
4. Peptide-Based Oligo-Functional Macromers

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Properties | Photo-Degradability | cCL: Radical Polymerization | cCL—conj.: Azide—Alkyne | cCL—conj: Diels-Alder | cCL—conj.: Thiol-Ene, Michael Addition |
|---|---|---|---|---|---|
| Enzyme degradability | [137] | [117] | [104,137] | [138] | [104,139] |
| Cell adhesive domains | - | [117] | [137] | [138] | - |
| cCL—conj: Diels-Alder | - | - | - | - | [138] |

5. Macromers Based on ECM-Molecules or Biologically Active non-ECM Polysaccharides
5.1. Hyaluronic Acid (HA)-Based Macromers
5.2. Gelatin-Based Macromers
5.3. Gellan Gum (GG)-Based Macromers
5.4. Chitosan-Based Macromers
6. Conclusions
| Properties | Specific Degradability | Cell Adhesive Domains | pCL: Hydrogen Bonding | pCL: Ionic Cross-Linking | cCL: Radical Polymerization | cCL—conj.: Azide-Alkyne | cCL—conj: Diels-Alder | cCL—conj.: Thiol-ene, Michael Addition | cCL—conj.: Amide/Amine Formation | cCL—Other Mechanism | Other | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pCL: stimulus-responsive, hydrophobic effect | NiPAAm-based, other acrylamides | - | [97] | - | - | [52,53,62] | [70] | [68,69] | [36,42] | [56,57,58,59,60,64,94] | [50,74] | [50,55,56,62,92,94,95,96,99] |
| - | block copolymers | [125] | - | - | - | [78,79,80,81,85,86,87] | - | - | [35,83] | - | [82] | - |
| Poly (organophos-phazene)s (POP) | - | - | - | - | [91] | - | - | [89,90] | - | - | [91] | |
| cCL: radical polymerization | [126] | - | - | - | - | - | - | [102] | - | - | [32,102,123,124] | |
| Photo-degradability | - | - | - | - | [114,127] | [130,165] | - | [128,129] | - | - | [114] | |
| Cell adhesive domains | [139] | - | - | - | [88,107,113] | - | - | [115,139] | - | - | [107] | |
| ECM molecule | [165] | - | - | - | [113,122,162] | [165] | - | [161] | - | [161] | [122,161,162] | |
| Bioactive non-ECM polysaccharide | - | - | - | [168] | [168,169,175] | - | - | - | - | - | - | |
| Shape-memory gels | - | - | [136] | - | [136] | - | - | - | - | - | - | |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Drury, J.L.; Mooney, D.J. Hydrogels for tissue engineering: Scaffold design variables and applications. Biomaterials 2003, 24, 4337–4351. [Google Scholar] [CrossRef]
- DeForest, C.A.; Anseth, K.S. Advances in bioactive hydrogels to probe and direct cell fate. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 421–444. [Google Scholar] [CrossRef] [PubMed]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th anniversary article: Rational design and applications of hydrogels in regenerative medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Designing degradable hydrogels for orthogonal control of cell microenvironments. Chem. Soc. Rev. 2013, 42, 7335–7372. [Google Scholar] [CrossRef] [PubMed]
- Knipe, J.M.; Peppas, N.A. Multi-responsive hydrogels for drug delivery and tissue engineering applications. Regen. Biomater. 2014, 1, 57–65. [Google Scholar] [CrossRef]
- Malafaya, P.B.; Silva, G.A.; Reis, R.L. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv. Drug Deliv. Rev. 2007, 59, 207–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, H.L.; Hwang, Y.; Kar, M.; Varghese, S. Smart hydrogels as functional biomimetic systems. Biomater. Sci. 2014, 2, 603–618. [Google Scholar] [CrossRef]
- Tabata, Y. Biomaterial technology for tissue engineering applications. J. R. Soc. Interface 2009, 6, S311–S324. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.-H.; Ovsianikov, A.; Stampfl, J.; Liska, R. Additive manufacturing of photosensitive hydrogels for tissue engineering applications. BioNanoMaterials 2014, 15, 49–70. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Liu, W. Dipole-dipole and H-bonding interactions significantly enhance the multifaceted mechanical properties of thermoresponsive shape memory hydrogels. Adv. Funct. Mater. 2015, 25, 471–480. [Google Scholar] [CrossRef]
- Tamura, M.; Yanagawa, F.; Sugiura, S.; Takagi, T.; Sumaru, K.; Kanamori, T. Click-crosslinkable and photodegradable gelatin hydrogels for cytocompatible optical cell manipulation in natural environment. Sci. Rep. 2015, 5, 15060. [Google Scholar] [CrossRef] [PubMed]
- Galaev, I.; Mattiasson, B. Smart Polymers for Bioseparation and Bioprocessing; CRC Press: Boca Raton, FL, USA, 2001. [Google Scholar]
- Nič, M.; Jirát, J.; Košata, B.; Jenkins, A.; McNaught, A. IUPAC Compendium of Chemical Terminology; IUPAC: Research Triagle Park, NC, USA, 2009. [Google Scholar]
- Discher, D.E.; Mooney, D.J.; Zandstra, P.W. Growth factors, matrices, and forces combine and control stem cells. Science 2009, 324, 1673–1677. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Hilt, J.Z.; Khademhosseini, A.; Langer, R. Hydrogels in biology and medicine: From molecular principles to bionanotechnology. Adv. Mater. 2006, 18, 1345–1360. [Google Scholar] [CrossRef]
- Caló, E.; Khutoryanskiy, V.V. Biomedical applications of hydrogels: A review of patents and commercial products. Eur. Polym. J. 2015, 65, 252–267. [Google Scholar] [CrossRef]
- Kopeček, J. Hydrogel biomaterials: A smart future? Biomaterials 2007, 28, 5185–5192. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, M.; Smeets Niels, M.B.; Hoare, T. Designing injectable, covalently cross-linked hydrogels for biomedical applications. Macromol. Rapid Commun. 2014, 35, 598–617. [Google Scholar] [CrossRef] [PubMed]
- Chaterji, S.; Kwon, I.K.; Park, K. Smart polymeric gels: Redefining the limits of biomedical devices. Progr. Polym. Sci. 2007, 32, 1083–1122. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.; Orme, R.P.; Fricker, R.A.; Roach, P. Remote and local control of stimuli responsive materials for therapeutic applications. Adv. Drug Deliv. Rev. 2013, 65, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Peppas, N.A.; Bures, P.; Leobandung, W.; Ichikawa, H. Hydrogels in pharmaceutical formulations. Eur. J. Pharm. Biopharm. 2000, 50, 27–46. [Google Scholar] [CrossRef]
- Qiu, Y.; Park, K. Environment-sensitive hydrogels for drug delivery. Adv. Drug Deliv. Rev. 2001, 53, 321–339. [Google Scholar] [CrossRef]
- Rzaev, Z.M.O.; Dinçer, S.; Pişkin, E. Functional copolymers of N-isopropylacrylamide for bioengineering applications. Progr. Polym. Sci. 2007, 32, 534–595. [Google Scholar] [CrossRef]
- Hennink, W.E.; van Nostrum, C.F. Novel crosslinking methods to design hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 13–36. [Google Scholar] [CrossRef]
- Nguyen, Q.V.; Huynh, D.P.; Park, J.H.; Lee, D.S. Injectable polymeric hydrogels for the delivery of therapeutic agents: A review. Eur. Polym. J. 2015. [Google Scholar] [CrossRef]
- Hoffman, A.S. Hydrogels for biomedical applications. Adv. Drug Deliv. Rev. 2002, 54, 3–12. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, J.; Deng, C.; Suuronen, E.J.; Zhong, Z. Click hydrogels, microgels and nanogels: Emerging platforms for drug delivery and tissue engineering. Biomaterials 2014, 35, 4969–4985. [Google Scholar] [CrossRef] [PubMed]
- Hacker, M.C.; Mikos, A.G. Chapter 33—Synthetic polymers. In Principles of Regenerative Medicine, 2nd ed.; Atala, A., Lanza, R., Thomson, J., Nerem, R.M., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 587–622. [Google Scholar]
- Husár, B.; Liska, R. Vinyl carbonates, vinyl carbamates, and related monomers: Synthesis, polymerization, and application. Chem. Soc. Rev. 2012, 41, 2395–2405. [Google Scholar] [CrossRef] [PubMed]
- Kasper, F.K.; Tanahashi, K.; Fisher, J.P.; Mikos, A.G. Synthesis of poly(propylene fumarate). Nat. Protoc. 2009, 4, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Kinard, L.A.; Kasper, F.K.; Mikos, A.G. Synthesis of oligo(poly(ethylene glycol) fumarate). Nat. Protoc. 2012, 7, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, D.; Nair, P.; Kim, H.K.W.; Yang, J. Citrate-based biodegradable injectable hydrogel composites for orthopedic applications. Biomater. Sci. 2013, 1, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Mather, B.D.; Viswanathan, K.; Miller, K.M.; Long, T.E. Michael addition reactions in macromolecular design for emerging technologies. Progr. Polymer Sci. 2006, 31, 487–531. [Google Scholar] [CrossRef]
- Nimmo, C.M.; Shoichet, M.S. Regenerative biomaterials that “click”: Simple, aqueous-based protocols for hydrogel synthesis, surface immobilization, and 3D patterning. Bioconjug. Chem. 2011, 22, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Lee, J.S.; Webb, K. Formulation and characterization of poloxamine-based hydrogels as tissue sealants. Acta Biomater. 2012, 8, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Bearat, H.H.; Lee, B.H.; Vernon, B.L. Comparison of properties between NiPAAm-based simultaneously physically and chemically gelling polymer systems for use in vivo. Acta Biomater. 2012, 8, 3629–3642. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.; Kim, S.W.; Bae, Y.H. Thermosensitive sol-gel reversible hydrogels. Adv. Drug Deliv. Rev. 2002, 54, 37–51. [Google Scholar] [CrossRef]
- Schild, H.G.; Tirrell, D.A. Microcalorimetric detection of lower critical solution temperatures in aqueous polymer solutions. J. Phys. Chem. 1990, 94, 4352–4356. [Google Scholar] [CrossRef]
- Pelton, R. Temperature-sensitive aqueous microgels. Adv. Colloid Interface Sci. 2000, 85, 1–33. [Google Scholar] [CrossRef]
- Cao, Y.; Zhu, X.X.; Luo, J.; Liu, H. Effects of substitution groups on the RAFT polymerization of N-alkylacrylamides in the preparation of thermosensitive block copolymers. Macromolecules 2007, 40, 6481–6488. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Gibson, M.I.; Klok, H.A. Synthese funktioneller Polymere durch polymeranaloge Reaktionen. Angew. Chem. 2009, 121, 50–60. [Google Scholar] [CrossRef]
- Robb, S.A.; Lee, B.H.; McLemore, R.; Vernon, B.L. Simultaneously physically and chemically gelling polymer system utilizing a poly(NiPAAm-co-cysteamine)-based copolymer. Biomacromolecules 2007, 8, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L.; Hubbell, J.A. Conjugate addition reactions combined with free-radical cross-linking for the design of materials for tissue engineering. Biomacromolecules 2001, 2, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Elbert, D.L.; Pratt, A.B.; Lutolf, M.P.; Halstenberg, S.; Hubbell, J.A. Protein delivery from materials formed by self-selective conjugate addition reactions. J. Control. Release 2001, 76, 11–25. [Google Scholar] [CrossRef]
- Fairbanks, B.D.; Schwartz, M.P.; Halevi, A.E.; Nuttelman, C.R.; Bowman, C.N.; Anseth, K.S. A Versatile synthetic extracellular matrix mimic via thiol-norbornene photopolymerization. Adv. Mater. 2009, 21, 5005–5010. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthesis and physicochemical characterization of end-linked poly(ethylene glycol)-co-peptide hydrogels formed by Michael-type addition. Biomacromolecules 2003, 4, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Hoyle, C.E.; Bowman, C.N. Thiol–ene click chemistry. Angew. Chem. Int. Ed. 2010, 49, 1540–1573. [Google Scholar] [CrossRef] [PubMed]
- Nair, D.P.; Podgórski, M.; Chatani, S.; Gong, T.; Xi, W.; Fenoli, C.R.; Bowman, C.N. The thiol-michael addition click reaction: A powerful and widely used tool in materials chemistry. Chem. Mater. 2014, 26, 724–744. [Google Scholar] [CrossRef]
- Patenaude, M.; Campbell, S.; Kinio, D.; Hoare, T. Tuning gelation time and morphology of injectable hydrogels using ketone-hydrazide cross-linking. Biomacromolecules 2014, 15, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Patenaude, M.; Hoare, T. Injectable, degradable thermoresponsive poly(N-isopropylacrylamide) hydrogels. ACS Macro Lett. 2012, 1, 409–413. [Google Scholar] [CrossRef]
- Hacker, M.C.; Klouda, L.; Ma, B.B.; Kretlow, J.D.; Mikos, A.G. Synthesis and characterization of injectable, thermally and chemically gelable, amphiphilic poly(N-isopropylacrylamide)-based macromers. Biomacromolecules 2008, 9, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Klouda, L.; Hacker, M.C.; Kretlow, J.D.; Mikos, A.G. Cytocompatibility evaluation of amphiphilic, thermally responsive and chemically crosslinkable macromers for in situ forming hydrogels. Biomaterials 2009, 30, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Klouda, L.; Perkins, K.R.; Watson, B.M.; Hacker, M.C.; Bryant, S.J.; Raphael, R.M.; Kurtis Kasper, F.; Mikos, A.G. Thermoresponsive, in situ cross-linkable hydrogels based on N-isopropylacrylamide: Fabrication, characterization and mesenchymal stem cell encapsulation. Acta Biomater. 2011, 7, 1460–1467. [Google Scholar] [CrossRef] [PubMed]
- Kretlow, J.D.; Hacker, M.C.; Klouda, L.; Ma, B.B.; Mikos, A.G. Synthesis and characterization of dual stimuli responsive macromers based on poly(N-isopropylacrylamide) and poly(vinylphosphonic acid). Biomacromolecules 2010, 11, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Loth, T.; Hennig, R.; Kascholke, C.; Hötzel, R.; Hacker, M.C. Reactive and stimuli-responsive maleic anhydride containing macromers—Multi-functional cross-linkers and building blocks for hydrogel fabrication. React. Funct. Polym. 2013, 73, 1480–1492. [Google Scholar] [CrossRef]
- Loth, T.; Hötzel, R.; Kascholke, C.; Anderegg, U.; Schulz-Siegmund, M.; Hacker, M.C. Gelatin-based biomaterial engineering with anhydride-containing oligomeric cross-linkers. Biomacromolecules 2014, 15, 2104–2118. [Google Scholar] [CrossRef] [PubMed]
- Ekenseair, A.K.; Boere, K.W.M.; Tzouanas, S.N.; Vo, T.N.; Kasper, F.K.; Mikos, A.G. Synthesis and characterization of thermally and chemically gelling injectable hydrogels for tissue engineering. Biomacromolecules 2012, 13, 1908–1915. [Google Scholar] [CrossRef] [PubMed]
- Ekenseair, A.K.; Boere, K.W.M.; Tzouanas, S.N.; Vo, T.N.; Kasper, F.K.; Mikos, A.G. Structure-property evaluation of thermally and chemically gelling injectable hydrogels for tissue engineering. Biomacromolecules 2012, 13, 2821–2830. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.N.; Ekenseair, A.K.; Kasper, F.K.; Mikos, A.G. Synthesis, physicochemical characterization, and cytocompatibility of bioresorbable, dual-gelling injectable hydrogels. Biomacromolecules 2014, 15, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.N.; Ekenseair, A.K.; Spicer, P.P.; Watson, B.M.; Tzouanas, S.N.; Roh, T.T.; Mikos, A.G. In vitro and in vivo evaluation of self-mineralization and biocompatibility of injectable, dual-gelling hydrogels for bone tissue engineering. J. Control. Release 2015, 205, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.M.; Kasper, F.K.; Engel, P.S.; Mikos, A.G. Synthesis and characterization of injectable, biodegradable, phosphate-containing, chemically cross-linkable, thermoresponsive macromers for bone tissue engineering. Biomacromolecules 2014, 15, 1788–1796. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.M.; Vo, T.N.; Tatara, A.M.; Shah, S.R.; Scott, D.W.; Engel, P.S.; Mikos, A.G. Biodegradable, phosphate-containing, dual-gelling macromers for cellular delivery in bone tissue engineering. Biomaterials 2015, 67, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Hong, Y.; Ma, Z.; Wagner, W.R. Protein-reactive, thermoresponsive copolymers with high flexibility and biodegradability. Biomacromolecules 2008, 9, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Hörner, S.; Uth, C.; Avrutina, O.; Frauendorf, H.; Wiessler, M.; Kolmar, H. Combination of inverse electron-demand Diels-Alder reaction with highly efficient oxime ligation expands the toolbox of site-selective peptide conjugations. Chem. Commun. 2015, 51, 11130–11133. [Google Scholar] [CrossRef] [PubMed]
- Hassert, R.; Pagel, M.; Ming, Z.; Häupl, T.; Abel, B.; Braun, K.; Wiessler, M.; Beck-Sickinger, A.G. Biocompatible silicon surfaces through orthogonal click chemistries and a high affinity silicon oxide binding peptide. Bioconjug. Chem. 2012, 23, 2129–2137. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Zeglis, B.M.; Lewis, J.S.; Anderson, C.J. The growing impact of bioorthogonal click chemistry on the development of radiopharmaceuticals. J. Nucl. Med. 2013, 54, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.-L.; Yang, Z.; Chu, H.-J.; Zhu, J.; Li, Z.-C.; Cui, J.-S. Facile preparation of poly(N-isopropylacrylamide)-based hydrogels via aqueous Diels-Alder click reaction. Polymer 2010, 51, 1694–1702. [Google Scholar] [CrossRef]
- Wei, H.-L.; Yang, Z.; Zheng, L.-M.; Shen, Y.-M. Thermosensitive hydrogels synthesized by fast Diels-Alder reaction in water. Polymer 2009, 50, 2836–2840. [Google Scholar] [CrossRef]
- Xu, X.; Chen, C.; Wang, Z.; Wang, G.; Cheng, S.; Zhang, X.; Zhuo, R. “Click” chemistry for in situ formation of thermoresponsive P(NiPAAm-co-HEMA)-based hydrogels. J. Polym. Sci. A Polym. Chem. 2008, 46, 5263–5277. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.-D.; Wu, D.-Q.; Zhang, X.-Z.; Zhuo, R.-X. Synthesis of thermosensitive P(NiPAAm-co-HEMA)/cellulose hydrogels via “click” chemistry. Carbohydr. Polym. 2009, 77, 583–589. [Google Scholar] [CrossRef]
- Xu, X.-D.; Chen, C.-S.; Lu, B.; Wang, Z.-C.; Cheng, S.-X.; Zhang, X.-Z.; Zhuo, R.-X. Modular synthesis of thermosensitive P(NiPAAm-co-HEMA)/β-CD based hydrogels via click chemistry. Macromol. Rapid Commun. 2009, 30, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.-H.; Su, J.; Messersmith, P.B. Hydrogels cross-linked by native chemical ligation. Biomacromolecules 2009, 10, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Boere Kristel, W.M.; van den Dikkenberg, J.; Gao, Y.; Visser, J.; Hennink, W.E.; Vermonden, T. Thermogelling and chemoselectively cross-linked hydrogels with controlled mechanical properties and degradation behavior. Biomacromolecules 2015, 16, 2840–2851. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.; Muir, T.; Clark-Lewis, I.; Kent, S. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Boere, K.W.M.; Soliman, B.G.; Rijkers, Dirk T. S.; Hennink, W.E.; Vermonden, T. Thermoresponsive injectable hydrogels cross-linked by native chemical ligation. Macromolecules 2014, 47, 2430–2438. [Google Scholar] [CrossRef]
- Wan, Q.; Chen, J.; Yuan, Y.; Danishefsky, S.J. Oxo-ester mediated native chemical ligation: Concept and applications. J. Am. Chem. Soc. 2008, 130, 15814–15816. [Google Scholar] [CrossRef] [PubMed]
- Di Biase, M.; Saunders, R.E.; Tirelli, N.; Derby, B. Inkjet printing and cell seeding thermoreversible photocurable gel structures. Soft Matter 2011, 7, 2639–2646. [Google Scholar] [CrossRef]
- Sosnik, A.; Cohn, D.; San Roman, J.; Abraham, G.A. Crosslinkable PEO-PPO-PEO-based reverse thermo-responsive gels as potentially injectable materials. J. Biomater. Sci. Polym. Ed. 2003, 14, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Swennen, I.; Vermeersch, V.; Hornof, M.; Adriaens, E.; Remon, J.-P.; Urtti, A.; Schacht, E.H. In-situ crosslinkable thermo-responsive hydrogels for drug delivery. J. Control. Release 2006, 116, e21–e24. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, N.E.; Swennen, I.; Girones, J.; Moroni, L.; van Blitterswijk, C.A.; Schacht, E.; Alblas, J.; Dhert, W.J.A. Evaluation of photocrosslinked Lutrol hydrogel for tissue printing applications. Biomacromolecules 2009, 10, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Cohn, D. Ethoxysilane-capped PEO-PPO-PEO triblocks: A new family of reverse thermo-responsive polymers. Biomaterials 2004, 25, 2851–2858. [Google Scholar] [CrossRef] [PubMed]
- Cellesi, F.; Tirelli, N.; Hubbell, J.A. Towards a fully-synthetic substitute of alginate: Development of a new process using thermal gelation and chemical cross-linking. Biomaterials 2004, 25, 5115–5124. [Google Scholar] [CrossRef] [PubMed]
- Cellesi, F.; Tirelli, N.; Hubbell, J.A. Materials for cell encapsulation via a new tandem approach combining reverse thermal gelation and covalent crosslinking. Macromol. Chem. Phys. 2002, 203, 1466–1472. [Google Scholar] [CrossRef]
- Fisher, J.P.; Jo, S.; Mikos, A.G.; Reddi, A.H. Thermoreversible hydrogel scaffolds for articular cartilage engineering. J. Biomed. Mater. Res. A 2004, 71, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Behravesh, E.; Shung, A.K.; Jo, S.; Mikos, A.G. Synthesis and characterization of triblock copolymers of methoxy poly(ethylene glycol) and poly(propylene fumarate). Biomacromolecules 2002, 3, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Behravesh, E.; Jo, S.; Zygourakis, K.; Mikos, A.G. Synthesis of in situ cross-linkable macroporous biodegradable poly(propylene fumarate-co-ethylene glycol) hydrogels. Biomacromolecules 2002, 3, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Behravesh, E.; Mikos, A.G. Three-dimensional culture of differentiating marrow stromal osteoblasts in biomimetic poly(propylene fumarate-co-ethylene glycol)-based macroporous hydrogels. J. Biomed. Mater. Res. A 2003, 66, 698–706. [Google Scholar] [CrossRef] [PubMed]
- Potta, T.; Chun, C.; Song, S.-C. Chemically crosslinkable thermosensitive polyphosphazene gels as injectable materials for biomedical applications. Biomaterials 2009, 30, 6178–6192. [Google Scholar] [CrossRef] [PubMed]
- Potta, T.; Chun, C.; Song, S.-C. Injectable, dual cross-linkable polyphosphazene blend hydrogels. Biomaterials 2010, 31, 8107–8120. [Google Scholar] [CrossRef] [PubMed]
- Nichol, J.L.; Allcock, H.R. Polyphosphazenes with amino acid citronellol ester side groups for biomedical applications. Eur. Polym. J. 2015, 62, 214–221. [Google Scholar] [CrossRef]
- Lin, Z.; Cao, S.; Chen, X.; Wu, W.; Li, J. Thermoresponsive hydrogels from phosphorylated ABA triblock copolymers: A potential scaffold for bone tissue engineering. Biomacromolecules 2013, 14, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Watson, B.M.; Kasper, F.K.; Mikos, A.G. Phosphorous-containing polymers for regenerative medicine. Biomed. Mater. 2014, 9, 25014. [Google Scholar] [CrossRef]
- Fathi, A.; Mithieux, S.M.; Wei, H.; Chrzanowski, W.; Valtchev, P.; Weiss, A.S.; Dehghani, F. Elastin based cell-laden injectable hydrogels with tunable gelation, mechanical and biodegradation properties. Biomaterials 2014, 35, 5425–5435. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, Z.; Li, X.; Fan, Z.; Liu, Z.; Xie, X.; Guan, J. Regulating myogenic differentiation of mesenchymal stem cells using thermosensitive hydrogels. Acta Biomater. 2015, 26, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; van Lith, R.; Baler, K.; Hoshi, R.A.; Ameer, G.A. A thermoresponsive biodegradable polymer with intrinsic antioxidant properties. Biomacromolecules 2014, 15, 3942–3952. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-H.; Kim, M.-H.; Park, S.-H.; Lee, H.J.; Kim, I.K.; Chung, H.-M. Synthesis of Arg-Gly-Asp (RGD) sequence conjugated thermo-reversible gel via the PEG spacer arm as an extracellular matrix for a pheochromocytoma cell (PC12) culture. Biosci. Biotechnol. Biochem. 2004, 68, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Na, K.; Kim, S.W.; Jung, S.Y.; Park, K.H.; Chung, H.M. Phenotype of hepatocyte spheroids behavior within thermo-sensitive poly(NiPAAm-co-PEG-g-GRGDS) hydrogel as a cell delivery vehicle. Biotechnol. Lett. 2005, 27, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Zhao, H.-B.; Yu, L.-X.; Chen, S.-C.; Wang, Y.-Z. Multi-stimuli sensitive supramolecular hydrogel formed by host–guest interaction between PNIPAM-Azo and cyclodextrin dimers. RSC Adv. 2014, 4, 4955. [Google Scholar] [CrossRef]
- Patterson, J.; Martino, M.M.; Hubbell, J.A. Biomimetic materials in tissue engineering. Mater. Today 2010, 13, 14–22. [Google Scholar] [CrossRef]
- Zhu, J. Bioactive modification of poly(ethylene glycol) hydrogels for tissue engineering. Biomaterials 2010, 31, 4639–4656. [Google Scholar] [CrossRef] [PubMed]
- Han, L.-H.; Tong, X.; Yang, F. Photo-crosslinkable PEG-based microribbons for forming 3D macroporous scaffolds with decoupled niche properties. Adv. Mater. 2014, 26, 1757–1762. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Smith Callahan, L.A.; Hao, J.; Guo, K.; Wesdemiotis, C.; Weiss, R.A.; Becker, M.L. Strain-promoted crosslinking of PEG-based hydrogels via Copper-free cycloaddition. ACS Macro Lett. 2012, 1, 1071–1073. [Google Scholar] [CrossRef] [PubMed]
- Deforest, C.A.; Polizzotti, B.D.; Anseth, K.S. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 2009, 8, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.T.; Marchesan, S.; Evans, R.A.; Styan, K.E.; Such, G.K.; Postma, A.; McLean, K.M.; Muir, B.W.; Caruso, F. Photoinitiated alkyne-azide click and radical cross-linking reactions for the patterning of PEG hydrogels. Biomacromolecules 2012, 13, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Deans, T.L.; Elisseeff, J.H. Stem cells in musculoskeletal engineered tissue. Curr. Opin. Biotechnol. 2009, 20, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Zhan, J.; Ye, Z.; Elisseeff, J.H. Modular multifunctional poly(ethylene glycol) hydrogels for stem cell differentiation. Adv. Funct. Mater. 2013, 23, 575–582. [Google Scholar] [CrossRef]
- Hench, L.L.; Polak, J.M. Third-generation biomedical materials. Science 2002, 295, 1014–1017. [Google Scholar] [CrossRef] [PubMed]
- Bellis, S.L. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials 2011, 32, 4205–4210. [Google Scholar] [CrossRef] [PubMed]
- Hersel, U.; Dahmen, C.; Kessler, H. RGD modified polymers: Biomaterials for stimulated cell adhesion and beyond. Biomaterials 2003, 24, 4385–4415. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell. Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.; Geng, W.; Ito, T.; Kohane, D.S.; Burdick, J.A.; Radisic, M. Photocrosslinkable hydrogel for myocyte cell culture and injection. J. Biomed. Mater. Res. Part B 2007, 81, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.R.; Schlosser, J.L.; Lam, S.F.; Nguyen, T.H.; Maynard, H.D.; Kasko, A.M. Synthesis of photodegradable macromers for conjugation and release of bioactive molecules. Biomacromolecules 2013, 14, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Phelps, E.A.; Enemchukwu, N.O.; Fiore, V.F.; Sy, J.C.; Murthy, N.; Sulchek, T.A.; Barker, T.H.; Garcia, A.J. Maleimide cross-linked bioactive PEG hydrogel exhibits improved reaction kinetics and cross-linking for cell encapsulation and in situ delivery. Adv. Mater. 2012, 24, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.; Hubbell, J.A. Enhanced proteolytic degradation of molecularly engineered PEG hydrogels in response to MMP-1 and MMP-2. Biomaterials 2010, 31, 7836–7845. [Google Scholar] [CrossRef] [PubMed]
- Halstenberg, S.; Panitch, A.; Rizzi, S.; Hall, H.; Hubbell, J.A. Biologically engineered protein-graft-poly(ethylene glycol) hydrogels: A cell adhesive and plasmin-degradable biosynthetic material for tissue repair. Biomacromolecules 2002, 3, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Lutolf, M.P.; Hubbell, J.A. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat. Biotechnol. 2005, 23, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rodrigues, J.; Tomas, H. Injectable and biodegradable hydrogels: Gelation, biodegradation and biomedical applications. Chem. Soc. Rev. 2012, 41, 2193–2221. [Google Scholar] [CrossRef] [PubMed]
- Petit, A.; Muller, B.; Bruin, P.; Meyboom, R.; Piest, M.; Batenburg, K.; Loes, M.J.; Leo, G.J.; Hennink, W.E.; Vermonden, T. Modulating rheological and degradation properties of temperature-responsive gelling systems composed of blends of PCLA-PEG-PCLA triblock copolymers and their fully hexanoyl-capped derivatives. Acta Biomater. 2012, 8, 4260–4267. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Chung, C.; Khetan, S.; Burdick, J.A. Hydrolytically degradable hyaluronic acid hydrogels with controlled temporal structures. Biomacromolecules 2008, 9, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-A.; Williams, C.G.; Li, Q.; Sharma, B.; Elisseeff, J.H. Synthesis and characterization of a novel degradable phosphate-containing hydrogel. Biomaterials 2003, 24, 3969–3980. [Google Scholar] [CrossRef]
- Li, Q.; Wang, J.; Shahani, S.; Sun, D.D.N.; Sharma, B.; Elisseeff, J.H.; Leong, K.W. Biodegradable and photocrosslinkable polyphosphoester hydrogel. Biomaterials 2006, 27, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.H.; Sohn, Y.S.; Jeong, B. Thermogelling poly(ethylene oxide-b-propylene oxide-b-ethylene oxide) disulfide multiblock copolymer as a thiol-sensitive degradable polymer. Biomacromolecules 2006, 7, 2871–2877. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Shen, C.J.; Legant, W.R.; Baranski, J.D.; Blakely, B.L.; Chen, C.S. Bioactive hydrogels made from step-growth derived PEG-peptide macromers. Biomaterials 2010, 31, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Griffin, D.R.; Kasko, A.M. Photodegradable macromers and hydrogels for live cell encapsulation and release. J. Am. Chem. Soc. 2012, 134, 13103–13107. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Kiick, K.L.; Kloxin, A.M. Design of thiol- and light-sensitive degradable hydrogels using Michael-type addition reactions. Polym. Chem. 2015, 6, 5565–5574. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.M.; Kloxin, A.M.; Kiick, K.L. Dually degradable click hydrogels for controlled degradation and protein release. J. Mater. Chem. B 2014, 2, 5511–5521. [Google Scholar] [CrossRef] [PubMed]
- Azagarsamy, M.A.; McKinnon, D.D.; Alge, D.L.; Anseth, K.S. Coumarin-based photodegradable hydrogel: Design, synthesis, gelation, and degradation kinetics. ACS Macro Lett. 2014, 3, 515–519. [Google Scholar] [CrossRef]
- Lendlein, A.; Kelch, S. Shape-memory polymers. Angew. Chem. Int. Ed. Engl. 2002, 41, 2035–2057. [Google Scholar] [CrossRef]
- Liu, C.; Qin, H.; Mather, P.T. Review of progress in shape-memory polymers. J. Mater. Chem. 2007, 17, 1543–1558. [Google Scholar] [CrossRef]
- Huang, W.M.; Song, C.L.; Fu, Y.Q.; Wang, C.C.; Zhao, Y.; Purnawali, H.; Lu, H.B.; Tang, C.; Ding, Z.; Zhang, J.L. Shaping tissue with shape memory materials. Adv. Drug Deliv. Rev. 2013, 65, 515–535. [Google Scholar] [CrossRef] [PubMed]
- Osada, Y.; Matsuda, A. Shape memory in hydrogels. Nature 1995, 376, 219. [Google Scholar] [CrossRef] [PubMed]
- Lendlein, A.; Jiang, H.; Jünger, O.; Langer, R. Light-induced shape-memory polymers. Nature 2005, 434, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Tan, M.; Cui, Y.; Deng, C.; Huang, H.; Guo, M. Reactive macromolecular micelle crosslinked highly elastic hydrogel with water-triggered shape-memory behaviour. Polym. Chem. 2014, 5, 4965–4973. [Google Scholar] [CrossRef]
- Kloxin, A.M.; Lewis, K.J.R.; DeForest, C.A.; Seedorf, G.; Tibbitt, M.W.; Balasubramaniam, V.; Anseth, K.S. Responsive culture platform to examine the influence of microenvironmental geometry on cell function in 3D. Integr. Biol. 2012, 4, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Alge, D.L.; Azagarsamy, M.A.; Donohue, D.F.; Anseth, K.S. Synthetically tractable click hydrogels for three-dimensional cell culture formed using tetrazine-norbornene chemistry. Biomacromolecules 2013, 14, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Tsurkan, M.V.; Chwalek, K.; Levental, K.R.; Freudenberg, U.; Werner, C. Modular starPEG-heparin gels with bifunctional peptide linkers. Macromol. Rapid Commun. 2010, 31, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- DeForest, C.A.; Anseth, K.S. Cytocompatible Click-based hydrogels with dynamically-tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011, 3, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Schurig, K.; Zieris, A.; Hermann, A.; Freudenberg, U.; Heidel, S.; Grimmer, M.; Storch, A.; Werner, C. Neurotropic growth factors and glycosaminoglycan based matrices to induce dopaminergic tissue formation. Biomaterials 2015, 67, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Zieris, A.; Dockhorn, R.; Rohrich, A.; Zimmermann, R.; Muller, M.; Welzel, P.B.; Tsurkan, M.V.; Sommer, J.-U.; Freudenberg, U.; Werner, C. Biohybrid networks of selectively desulfated glycosaminoglycans for tunable growth factor delivery. Biomacromolecules 2014, 15, 4439–4446. [Google Scholar] [CrossRef] [PubMed]
- Stabenfeldt, S.E.; García, A.J.; LaPlaca, M.C. Thermoreversible laminin-functionalized hydrogel for neural tissue engineering. J. Biomed. Mater. Res. A 2006, 77, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.J.; Zhu, Y.; Liu, F.S.; Nie, J.; Ma, J.; Yang, W.T. Comb-shaped conjugates comprising hydroxypropyl cellulose backbones and low-molecular-weight poly(N-isopropylacryamide) side chains for smart hydrogels: Synthesis, characterization, and biomedical applications. Bioconjug. Chem. 2010, 21, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.J.; Bae, J.W.; Park, H.D.; Lee, J.W.; Park, K.D. Thermosensitive chitosans as novel injectable biomaterials. Macromol. Symp. 2005, 224, 275–286. [Google Scholar] [CrossRef]
- Lü, S.; Li, B.; Ni, B.; Sun, Z.; Liu, M.; Wang, Q. Thermoresponsive injectable hydrogel for three-dimensional cell culture: Chondroitin sulfate bioconjugated with poly(N-isopropylacrylamide) synthesized by RAFT polymerization. Soft Matter 2011, 7, 10763–10772. [Google Scholar] [CrossRef]
- Ohya, S.; Sonoda, H.; Nakayama, Y.; Matsuda, T. The potential of poly(N-isopropylacrylamide) (PNIPAM)-grafted hyaluronan and PNIPAM-grafted gelatin in the control of post-surgical tissue adhesions. Biomaterials 2005, 26, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.; Ramirez, C.M.; Miljkovic, N.; Li, H.; Rubin, J.P.; Marra, K.G. Thermosensitive injectable hyaluronic acid hydrogel for adipose tissue engineering. Biomaterials 2009, 30, 6844–6853. [Google Scholar] [CrossRef] [PubMed]
- Ibusuki, S.; Fujii, Y.; Iwamoto, Y.; Matsuda, T. Tissue-engineered cartilage using an injectable and in situ gelable thermoresponsive gelatin: Fabrication and in vitro performance. Tissue Eng. 2003, 9, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, D.; Chu, C.-C. Synthesis and characterization of partially biodegradable, temperature and pH sensitive Dex-MA/PNIPAAm hydrogels. Biomaterials 2004, 25, 4719–4730. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Matsuda, T. Poly(N-isopropylacrylamide) (PNIPAM)-grafted gelatin as thermoresponsive three-dimensional artificial extracellular matrix: Molecular and formulation parameters vs. cell proliferation potential. J. Biomater. Sci. Polym. Ed. 2005, 16, 809–827. [Google Scholar] [CrossRef] [PubMed]
- Ibusuki, S.; Iwamoto, Y.; Matsuda, T. System-engineered cartilage using poly(N-isopropylacrylamide)-grafted gelatin as in situ-formable scaffold: in vivo performance. Tissue Eng. 2003, 9, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Ohya, S.; Nakayama, Y.; Matsuda, T. Thermoresponsive artificial extracellular matrix for tissue engineering: Hyaluronic acid bioconjugated with poly(N-isopropylacrylamide) grafts. Biomacromolecules 2001, 2, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Kogan, G.; Šoltés, L.; Stern, R.; Gemeiner, P. Hyaluronic acid: A natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol. Lett. 2007, 29, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Dicker, K.T.; Gurski, L.A.; Pradhan-Bhatt, S.; Witt, R.L.; Farach-Carson, M.C.; Jia, X. Hyaluronan: A simple polysaccharide with diverse biological functions. Acta Biomater. 2014, 10, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Anderegg, U.; Simon, J.C.; Averbeck, M. More than just a filler—The role of hyaluronan for skin homeostasis. Exp. Dermatol. 2014, 23, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Vigetti, D.; Karousou, E.; Viola, M.; Deleonibus, S.; Luca, G.D.; Passi, A. Hyaluronan: Biosynthesis and signaling. Biochim. Biophys. Acta 2014, 1840, 2452–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdick, J.A.; Prestwich, G.D. Hyaluronic acid hydrogels for biomedical applications. Adv. Mater. 2011, 23, H41–H56. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, G.D. Hyaluronic acid-based clinical biomaterials derived for cell and molecule delivery in regenerative medicine. J. Control. Release 2011, 155, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, G.D.; Erickson, I.E.; Zarembinski, T.I.; West, M.; Tew, W.P. The translational imperative: Making cell therapy simple and effective. Acta Biomater. 2012, 8, 4200–4207. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Chung, H.J.; Yeo, S.; Ahn, C.-H.; Lee, H.; Messersmith, P.B.; Park, T.G. Thermo-sensitive, injectable, and tissue adhesive sol-gel transition hyaluronic acid/pluronic composite hydrogels prepared from bio-inspired catechol-thiol reaction. Soft Matter 2010, 6, 977–983. [Google Scholar] [CrossRef]
- Purcell, B.P.; Kim, I.L.; Chuo, V.; Guinen, T.; Dorsey, S.M.; Burdick, J.A. Incorporation of sulfated hyaluronic acid macromers into degradable hydrogel scaffolds for sustained molecule delivery. Biomater. Sci. 2014, 2, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T. poly(N-isopropylacrylamide)-grafted gelatin as a thermoresponsive cell-adhesive, mold-releasable material for shape-engineered tissues. J. Biomater. Sci. Polym. Ed. 2004, 15, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Chan-Park, M.B. A biomimetic hydrogel based on methacrylated dextran-graft-lysine and gelatin for 3D smooth muscle cell culture. Biomaterials 2010, 31, 1158–1170. [Google Scholar] [CrossRef] [PubMed]
- Truong, V.X.; Tsang, K.M.; Simon, G.P.; Boyd, R.L.; Evans, R.A.; Thissen, H.; Forsythe, J.S. Photodegradable gelatin-based hydrogels prepared by bioorthogonal click chemistry for cell encapsulation and release. Biomacromolecules 2015, 16, 2246–2253. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Wang, C.; Lai, R.C.; Su, K.; Zhang, F.; Wang, D.-A. An improved injectable polysaccharide hydrogel: Modified gellan gum for long-term cartilage regeneration in vitro. J. Mater. Chem. 2009, 19, 1968–1977. [Google Scholar] [CrossRef]
- Oliveira, J.T.; Gardel, L.S.; Rada, T.; Martins, L.; Gomes, M.E.; Reis, R.L. Injectable gellan gum hydrogels with autologous cells for the treatment of rabbit articular cartilage defects. J. Orthop. Res. 2010, 28, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, D.F.; Sant, S.; Shin, H.; Oliveira, J.T.; Gomes, M.E.; Neves, N.M.; Khademhosseini, A.; Reis, R.L. Modified gellan gum hydrogels with tunable physical and mechanical properties. Biomaterials 2010, 31, 7494–7502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Correia, J.; Zavan, B.; Vindigni, V.; Silva, T.H.; Oliveira, J.M.; Abatangelo, G.; Reis, R.L. Biocompatibility evaluation of ionic- and photo-crosslinked methacrylated gellan gum hydrogels: In vitro and in vivo study. Adv. Healthc. Mater. 2013, 2, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Silva-Correia, J.; Oliveira, J.M.; Caridade, S.G.; Oliveira, J.T.; Sousa, R.A.; Mano, J.F.; Reis, R.L. Gellan gum-based hydrogels for intervertebral disc tissue-engineering applications. J. Tissue Eng. Regen. Med. 2011, 5, e97–e107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Correia, J.; Miranda-Goncalves, V.; Salgado, A.J.; Sousa, N.; Oliveira, J.M.; Reis, R.M.; Reis, R.L. Angiogenic potential of gellan-gum-based hydrogels for application in nucleus pulposus regeneration: In vivo study. Tissue Eng. Part A 2012, 18, 1203–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaryk, R.; Silva-Correia, J.; Oliveira, J.M.; Unger, R.E.; Landes, C.; Brochhausen, C.; Ghanaati, S.; Reis, R.L.; Kirkpatrick, C.J. Biological performance of cell-encapsulated methacrylated gellan gum-based hydrogels for nucleus pulposus regeneration. J. Tissue Eng. Regen. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Francis Suh, J.-K.; Matthew, H.W.T. Application of chitosan-based polysaccharide biomaterials in cartilage tissue engineering: A review. Biomaterials 2000, 21, 2589–2598. [Google Scholar] [CrossRef]
- Azuma, K.; Izumi, R.; Osaki, T.; Ifuku, S.; Morimoto, M.; Saimoto, H.; Minami, S.; Okamoto, Y. Chitin, chitosan, and its derivatives for wound healing: Old and new materials. J. Funct. Biomater. 2015, 6, 104–142. [Google Scholar] [CrossRef] [PubMed]
- Amsden, B.G.; Sukarto, A.; Knight, D.K.; Shapka, S.N. Methacrylated glycol chitosan as a photopolymerizable biomaterial. Biomacromolecules 2007, 8, 3758–3766. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hacker, M.C.; Nawaz, H.A. Multi-Functional Macromers for Hydrogel Design in Biomedical Engineering and Regenerative Medicine. Int. J. Mol. Sci. 2015, 16, 27677-27706. https://doi.org/10.3390/ijms161126056
Hacker MC, Nawaz HA. Multi-Functional Macromers for Hydrogel Design in Biomedical Engineering and Regenerative Medicine. International Journal of Molecular Sciences. 2015; 16(11):27677-27706. https://doi.org/10.3390/ijms161126056
Chicago/Turabian StyleHacker, Michael C., and Hafiz Awais Nawaz. 2015. "Multi-Functional Macromers for Hydrogel Design in Biomedical Engineering and Regenerative Medicine" International Journal of Molecular Sciences 16, no. 11: 27677-27706. https://doi.org/10.3390/ijms161126056
APA StyleHacker, M. C., & Nawaz, H. A. (2015). Multi-Functional Macromers for Hydrogel Design in Biomedical Engineering and Regenerative Medicine. International Journal of Molecular Sciences, 16(11), 27677-27706. https://doi.org/10.3390/ijms161126056