
The Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Attenuates Hepatic Lipogenesis via AMPK Activation in Non-Alcoholic Fatty Liver Disease Model Mice

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. General Observations

{kind=link}
{kind=link}
{kind=link}
| Measurement Item | Control | Teneligliptin |
|---|---|---|
| Body weight (g) | 83.4 ± 7.1 a | 80.7 ± 8.3 |
| Liver weight (g) | 5.5 ± 1.4 | 5.1 ± 0.8 |
| Liver-to-body weight ratio | 0.066 ± 0.013 | 0.063 ± 0.016 |
| White adipose tissue b (g) | 2.8 ± 0.7 | 2.8 ± 1.1 |
2.1.2. Effects of Teneligliptin on the Histopathology of the Experimental Mouse Liver

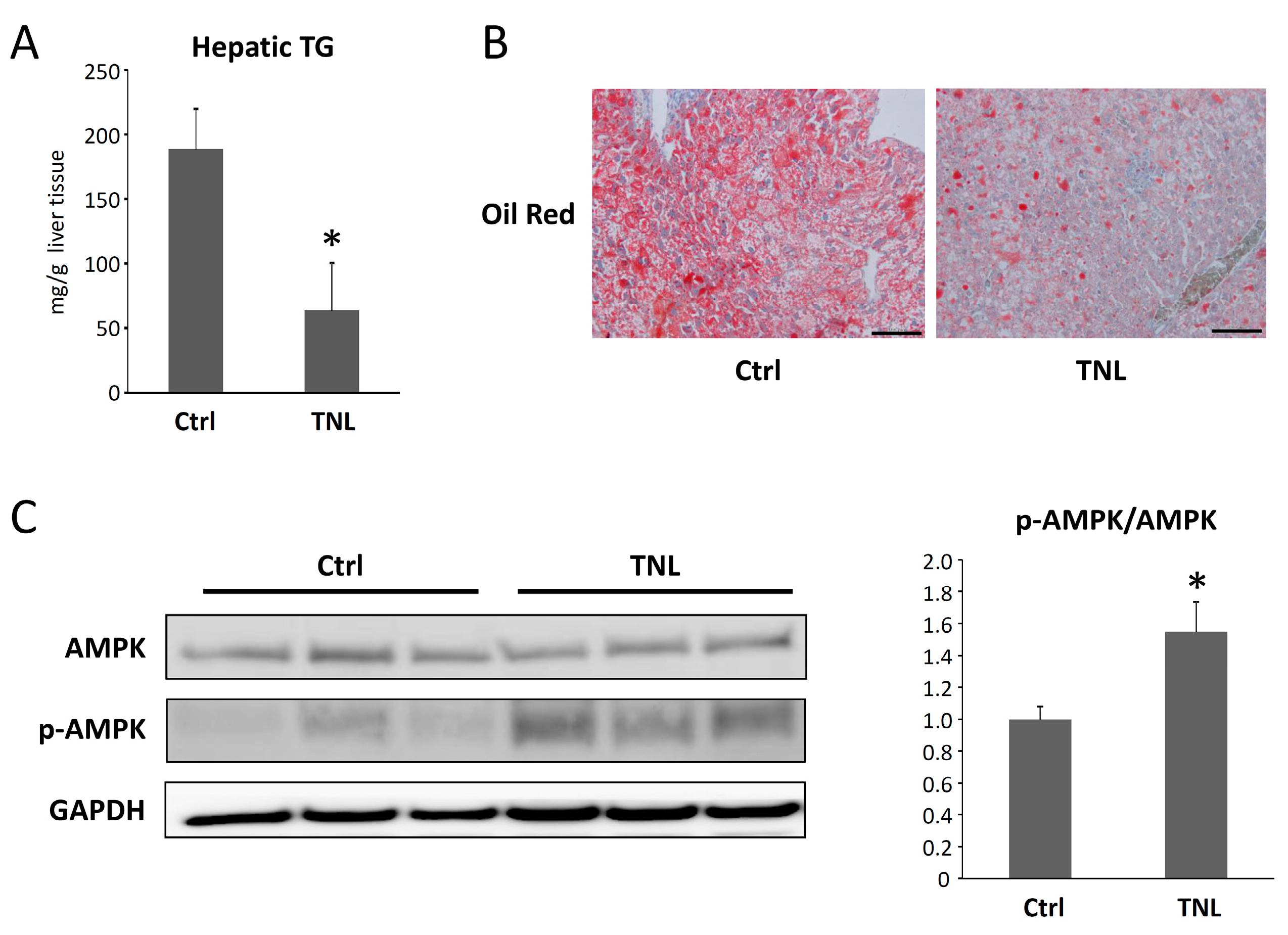
2.1.3. Effects of Teneligliptin on the Intrahepatic Triglyceride Levels and the Activation of AMP-Activated Protein Kinase in the Livers of Experimental Mice
2.1.4. Effects of Teneligliptin on the Expression Levels of Acetyl-CoA Carboxylase, Fatty Acid Synthetase, Sterol Regulatory Element-Binding Protein 1c and Elongation of Very Long Chain Fatty Acid-Like Family Member 6 mRNA in the Livers of Experimental Mice


2.1.5. Effects of Teneligliptin on Biochemical Parameters
| Measurement Item | Control | Teneligliptin |
|---|---|---|
| FFA (μEQ/mL) | 2091.0 ± 328.9 a | 1550.4 ± 267.5 |
| Glucose (mg/dL) | 295.2 ± 108.2 | 528.0 ± 102.0 |
| Insulin (ng/mL) | 2.3 ± 0.9 | 2.14 ± 1.8 |
| ALT (IU/L) | 239.8 ± 20.4 | 162.0 ± 16.5 b |
| Triglyceride (mg/mL) | 56.4 ± 32.2 | 65.2 ± 9.3 |
2.2. Discussion
3. Experimental Section
3.1. Animals and Chemicals
3.2. Experimental Procedure
3.3. Histopathological Examination
3.4. Clinical Chemistry
3.5. RNA Extraction and Quantitative Real-Time Reverse Transcription-PCR Analysis
| Genes | 5′-Primer | 3′-Primer |
|---|---|---|
| Acc | GGCTCAAACTGCAGGTATCC | TTGCCAATCCACTCGAAGA |
| Elovl6 | CAGCAAAGCACCCGAACTA | AGGAGCACAGTGATGTGGTG |
| Fas | GCTGCTGTTGGAAGTCAGC | AGTGTTCGTTCCTCGGAGTG |
| Srebp1c | CTGGAGCTGCGTGGTTT | GCCTCATGTAGGAATACCCTCCTCATA |
| 18s | CCATCCAATCGGTAGTAGCG | GTAACCCGTTGAACCCCATT |
3.6. Hepatic Lipid Analysis
3.7. Protein Extraction and Western Blot Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the american association for the study of liver diseases, american college of gastroenterology, and the american gastroenterological association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Sass, D.A.; Chang, P.; Chopra, K.B. Nonalcoholic fatty liver disease: A clinical review. Dig. Dis. Sci. 2005, 50, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar] [PubMed]
- Kim, C.H.; Younossi, Z.M. Nonalcoholic fatty liver disease: A manifestation of the metabolic syndrome. Clevel. Clin. J. Med. 2008, 75, 721–728. [Google Scholar]
- Vanni, E.; Bugianesi, E.; Kotronen, A.; De Minicis, S.; Yki-Jarvinen, H.; Svegliati-Baroni, G. From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis. 2010, 42, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, G.; Brizi, M.; Morselli-Labate, A.M.; Bianchi, G.; Bugianesi, E.; McCullough, A.J.; Forlani, G.; Melchionda, N. Association of nonalcoholic fatty liver disease with insulin resistance. Am. J. Med. 1999, 107, 450–455. [Google Scholar] [CrossRef]
- Sanyal, A.J.; American Gastroenterological, A. Aga technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Yasuda, Y.; Sakai, H.; Kubota, M.; Terakura, D.; Baba, A.; Ohno, T.; Kochi, T.; Tsurumi, H.; Tanaka, T.; et al. Pitavastatin suppresses diethylnitrosamine-induced liver preneoplasms in male C57BL/KsJ-db/db obese mice. BMC Cancer 2011, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Sakai, H.; Shirakami, Y.; Yasuda, Y.; Kubota, M.; Terakura, D.; Baba, A.; Ohno, T.; Hara, Y.; Tanaka, T.; et al. Preventive effects of (−)-epigallocatechin gallate on diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Prev. Res. 2011, 4, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Ohno, T.; Shimizu, M.; Shirakami, Y.; Baba, A.; Kochi, T.; Kubota, M.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Metformin suppresses diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-+Leprdb/+Leprdb mice. PLoS ONE 2015, 10, e0124081. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, T.; Zhang, C.; Tang, F.; Zhong, N.; Li, H.; Song, X.; Lin, H.; Liu, Y.; Xue, F. Identification of reciprocal causality between non-alcoholic fatty liver disease and metabolic syndrome by a simplified bayesian network in a chinese population. BMJ Open 2015, 5, e008204. [Google Scholar] [CrossRef] [PubMed]
- Aschner, P.; Kipnes, M.S.; Lunceford, J.K.; Sanchez, M.; Mickel, C.; Williams-Herman, D.E.; Sitagliptin Study, G. Effect of the dipeptidyl peptidase-4 inhibitor sitagliptin as monotherapy on glycemic control in patients with type 2 diabetes. Diabetes Care 2006, 29, 2632–2637. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, F.X.; Schweizer, A.; Mills, D.; Dejager, S. Efficacy and tolerability of vildagliptin monotherapy in drug-naive patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2007, 76, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like peptide-1 receptor agonism improves metabolic, biochemical, and histopathological indices of nonalcoholic steatohepatitis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Fujii, M.; Sandel, J.; Shibazaki, Y.; Wakamatsu, K.; Mark, M.; Yoneyama, H. Linagliptin alleviates hepatic steatosis and inflammation in a mouse model of non-alcoholic steatohepatitis. Med. Mol. Morphol. 2014, 47, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Balaban, Y.H.; Korkusuz, P.; Simsek, H.; Gokcan, H.; Gedikoglu, G.; Pinar, A.; Hascelik, G.; Asan, E.; Hamaloglu, E.; Tatar, G. Dipeptidyl peptidase IV (DDP IV) in nash patients. Ann. Hepatol. 2007, 6, 242–250. [Google Scholar] [PubMed]
- Schuppan, D.; Gorrell, M.D.; Klein, T.; Mark, M.; Afdhal, N.H. The challenge of developing novel pharmacological therapies for non-alcoholic steatohepatitis. Liver Int. 2010, 30, 795–808. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Y.; Atug, O.; Yonal, O.; Duman, D.; Ozdogan, O.; Imeryuz, N.; Kalayci, C. Dipeptidyl peptidase IV inhibitors: Therapeutic potential in nonalcoholic fatty liver disease. Med. Sci. Res. 2009, 15, HY1-5. [Google Scholar] [CrossRef]
- Collison, K.S.; Makhoul, N.J.; Zaidi, M.Z.; Al-Rabiah, R.; Inglis, A.; Andres, B.L.; Ubungen, R.; Shoukri, M.; Al-Mohanna, F.A. Interactive effects of neonatal exposure to monosodium glutamate and aspartame on glucose homeostasis. Nutr. Metabol. 2012, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Nagata, M.; Suzuki, W.; Iizuka, S.; Tabuchi, M.; Maruyama, H.; Takeda, S.; Aburada, M.; Miyamoto, K. Type 2 diabetes mellitus in obese mouse model induced by monosodium glutamate. Exp. Anim. Jpn. Assoc. Lab. Anim. Sci. 2006, 55, 109–115. [Google Scholar] [CrossRef]
- Roman-Ramos, R.; Almanza-Perez, J.C.; Garcia-Macedo, R.; Blancas-Flores, G.; Fortis-Barrera, A.; Jasso, E.I.; Garcia-Lorenzana, M.; Campos-Sepulveda, A.E.; Cruz, M.; Alarcon-Aguilar, F.J. Monosodium glutamate neonatal intoxication associated with obesity in adult stage is characterized by chronic inflammation and increased mRNA expression of peroxisome proliferator-activated receptors in mice. Basic Clin. Pharmacol. Toxicol. 2011, 108, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits Srebp activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metabol. 2011, 13, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Serviddio, G.; Bellanti, F.; Vendemiale, G. Free radical biology for medicine: Learning from nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2013, 65, 952–968. [Google Scholar] [CrossRef] [PubMed]
- Blaslov, K.; Bulum, T.; Zibar, K.; Duvnjak, L. Incretin based therapies: A novel treatment approach for non-alcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 7356–7365. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Sakai, H.; Shirakami, Y.; Iwasa, J.; Yasuda, Y.; Kubota, M.; Takai, K.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Acyclic retinoid inhibits diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BLKS/J- +leprdb/+leprdb mice. Cancer Prev. Res. 2011, 4, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein p, causes insulin resistance. Cell Metabol. 2010, 12, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.Y.; Hwang, S.Y.; Lee, C.H.; Hong, H.C.; Yang, S.J.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Increased selenoprotein p levels in subjects with visceral obesity and nonalcoholic fatty liver disease. Diabetes Metabol. J. 2013, 37, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Musso, G.; Gambino, R.; Cassader, M. Recent insights into hepatic lipid metabolism in non-alcoholic fatty liver disease (NAFLD). Progress Lipid Res. 2009, 48, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, N.; Kato, M.; Shundo, Y.; Tajiri, H.; Tanaka, M.; Yamashita, N.; Kohjima, M.; Kotoh, K.; Nakamuta, M.; Takayanagi, R.; et al. Liver X receptor in cooperation with Srebp-1c is a major lipid synthesis regulator in nonalcoholic fatty liver disease. Hepatol. Res. 2008, 38, 1122–1129. [Google Scholar] [CrossRef] [PubMed]
- Kern, M.; Kloting, N.; Niessen, H.G.; Thomas, L.; Stiller, D.; Mark, M.; Klein, T.; Bluher, M. Linagliptin improves insulin sensitivity and hepatic steatosis in diet-induced obesity. PLoS ONE 2012, 7, e38744. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Khera, R.; Allen, A.M.; Murad, M.H.; Loomba, R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: A systematic review and network meta-analysis. Hepatology 2015, 62, 1417–1432. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Shimizu, M.; Kubota, M.; Ohno, T.; Kochi, T.; Nakamura, N.; Sumi, T.; Tanaka, T.; Moriwaki, H.; Seishima, M. Pentoxifylline prevents nonalcoholic steatohepatitis-related liver pre-neoplasms by inhibiting hepatic inflammation and lipogenesis. Eur. J. Cancer Prev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Viollet, B.; Mounier, R.; Leclerc, J.; Yazigi, A.; Foretz, M.; Andreelli, F. Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes Metab. 2007, 33, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shlomo, S.; Zvibel, I.; Shnell, M.; Shlomai, A.; Chepurko, E.; Halpern, Z.; Barzilai, N.; Oren, R.; Fishman, S. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J. Hepatol. 2011, 54, 1214–1223. [Google Scholar] [CrossRef] [PubMed]
- Svegliati-Baroni, G.; Saccomanno, S.; Rychlicki, C.; Agostinelli, L.; de Minicis, S.; Candelaresi, C.; Faraci, G.; Pacetti, D.; Vivarelli, M.; Nicolini, D.; et al. Glucagon-like peptide-1 receptor activation stimulates hepatic lipid oxidation and restores hepatic signalling alteration induced by a high-fat diet in nonalcoholic steatohepatitis. Liver Int. 2011, 31, 1285–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hong, S.W.; Chae, S.W.; Kim, D.H.; Choi, J.H.; Bae, J.C.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; et al. Exendin-4 improves steatohepatitis by increasing Sirt1 expression in high-fat diet-induced obese C57BL/6J mice. PLoS ONE 2012, 7, e31394. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.L.; Bajaj, M. Potential of incretin-based therapies for non-alcoholic fatty liver disease. J. Diabetes Complicat. 2013, 27, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Boschmann, M.; Engeli, S.; Dobberstein, K.; Budziarek, P.; Strauss, A.; Boehnke, J.; Sweep, F.C.; Luft, F.C.; He, Y.; Foley, J.E.; et al. Dipeptidyl-peptidase-IV inhibition augments postprandial lipid mobilization and oxidation in type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2009, 94, 846–852. [Google Scholar] [CrossRef] [PubMed]
- The Japanese Association for Laboratory Animal Science (JALAS). Available online: http://www.jalas.jp/english/en_about_jalas.html (accessed on 26 July 2013).
- Shimizu, M.; Shirakami, Y.; Iwasa, J.; Shiraki, M.; Yasuda, Y.; Hata, K.; Hirose, Y.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Supplementation with branched-chain amino acids inhibits azoxymethane-induced colonic preneoplastic lesions in male C57BL/KsJ-db/db mice. Clin. Cancer Res. 2009, 15, 3068–3075. [Google Scholar] [CrossRef] [PubMed]
- Primer Blast. Available online: http://www.ncbi.nlm.nih.gov/tools/primer-blast/ (accessed on 26 July 2013).
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Iwasa, J.; Shimizu, M.; Shiraki, M.; Shirakami, Y.; Sakai, H.; Terakura, Y.; Takai, K.; Tsurumi, H.; Tanaka, T.; Moriwaki, H. Dietary supplementation with branched-chain amino acids suppresses diethylnitrosamine-induced liver tumorigenesis in obese and diabetic C57BL/KsJ-db/db mice. Cancer Sci. 2010, 101, 460–467. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, M.; Kato, M.; Tanaka, K.; Tanaka, M.; Kohjima, M.; Nakamura, K.; Enjoji, M.; Nakamuta, M.; Kotoh, K.; Takayanagi, R. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol. Med. Rep. 2012, 5, 729–733. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ideta, T.; Shirakami, Y.; Miyazaki, T.; Kochi, T.; Sakai, H.; Moriwaki, H.; Shimizu, M. The Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Attenuates Hepatic Lipogenesis via AMPK Activation in Non-Alcoholic Fatty Liver Disease Model Mice. Int. J. Mol. Sci. 2015, 16, 29207-29218. https://doi.org/10.3390/ijms161226156
Ideta T, Shirakami Y, Miyazaki T, Kochi T, Sakai H, Moriwaki H, Shimizu M. The Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Attenuates Hepatic Lipogenesis via AMPK Activation in Non-Alcoholic Fatty Liver Disease Model Mice. International Journal of Molecular Sciences. 2015; 16(12):29207-29218. https://doi.org/10.3390/ijms161226156
Chicago/Turabian StyleIdeta, Takayasu, Yohei Shirakami, Tsuneyuki Miyazaki, Takahiro Kochi, Hiroyasu Sakai, Hisataka Moriwaki, and Masahito Shimizu. 2015. "The Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Attenuates Hepatic Lipogenesis via AMPK Activation in Non-Alcoholic Fatty Liver Disease Model Mice" International Journal of Molecular Sciences 16, no. 12: 29207-29218. https://doi.org/10.3390/ijms161226156
APA StyleIdeta, T., Shirakami, Y., Miyazaki, T., Kochi, T., Sakai, H., Moriwaki, H., & Shimizu, M. (2015). The Dipeptidyl Peptidase-4 Inhibitor Teneligliptin Attenuates Hepatic Lipogenesis via AMPK Activation in Non-Alcoholic Fatty Liver Disease Model Mice. International Journal of Molecular Sciences, 16(12), 29207-29218. https://doi.org/10.3390/ijms161226156