
Combination Treatment with Sublethal Ionizing Radiation and the Proteasome Inhibitor, Bortezomib, Enhances Death-Receptor Mediated Apoptosis and Anti-Tumor Immune Attack


Abstract
: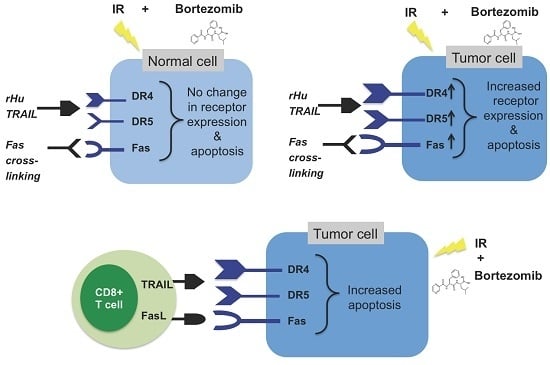
{kind=link}
{kind=link}
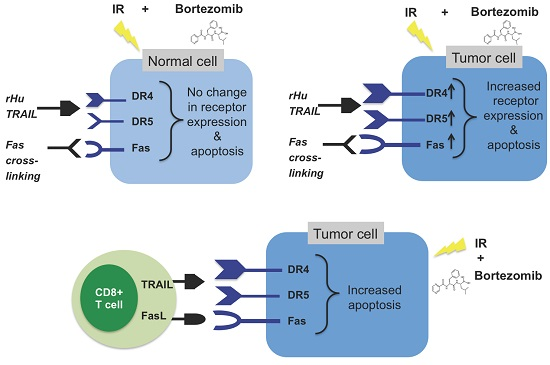
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effects of Combination Treatment on Colorectal Cancer Cell Viability

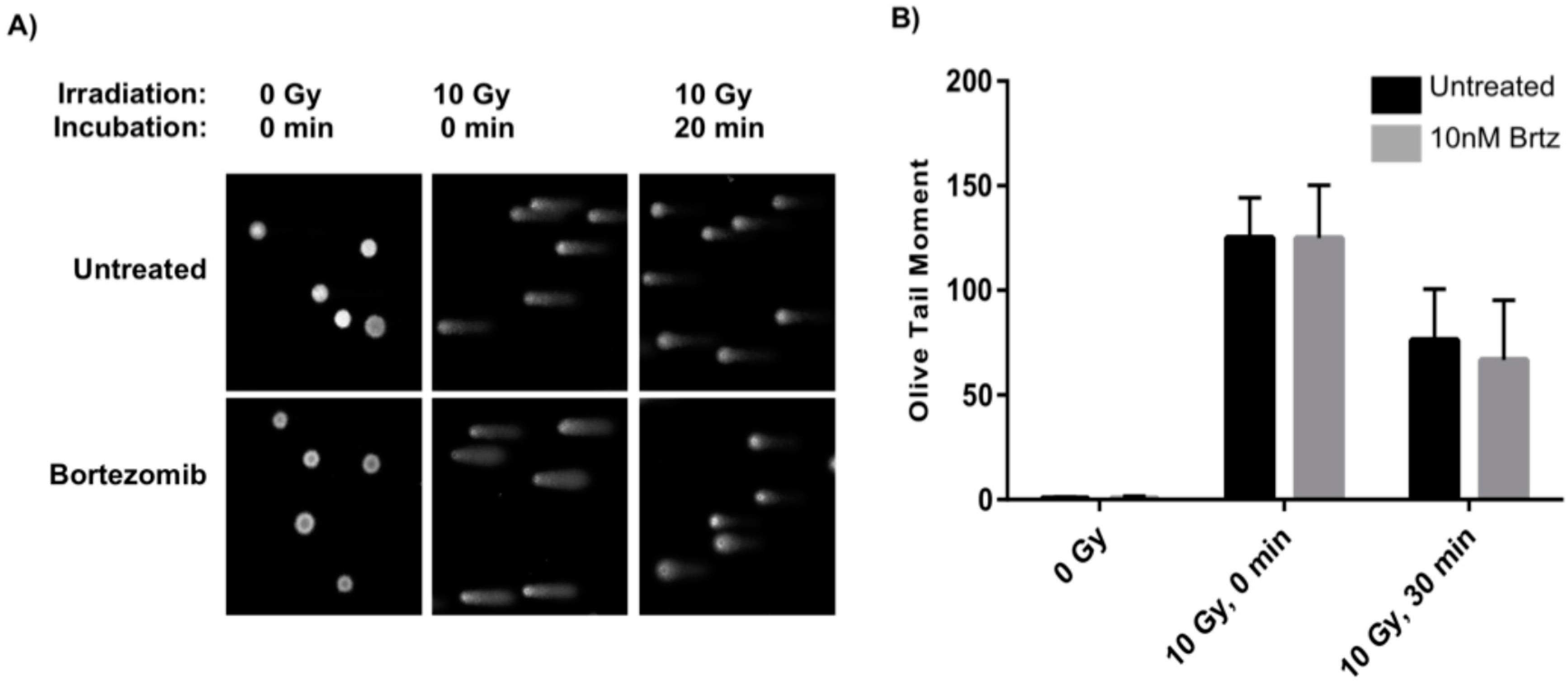
2.2. Combined Treatment Does Not Inhibit the Initial DNA Repair Response

2.3. Combination Treatment Further Enhances Transcript Expression of DR4, DR5 and Fas over Radiation or Inhibition of the 26S Proteasome Alone Treated Carcinoma Cells

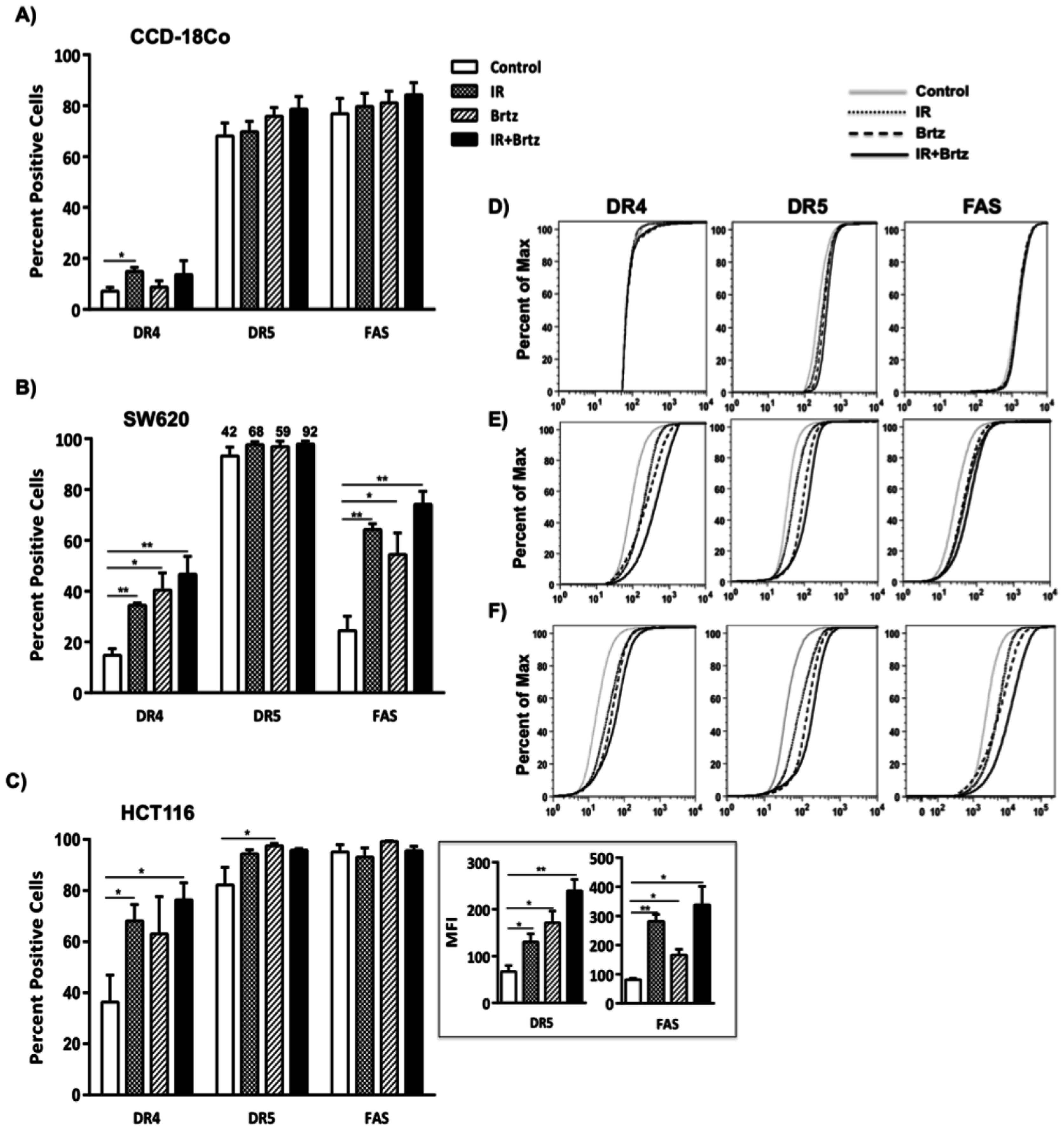
2.4. Combination Treatment with Bortezomib and Radiation Up-Regulates Cell Surface Protein Expression of Death Receptors in Tumor Cells
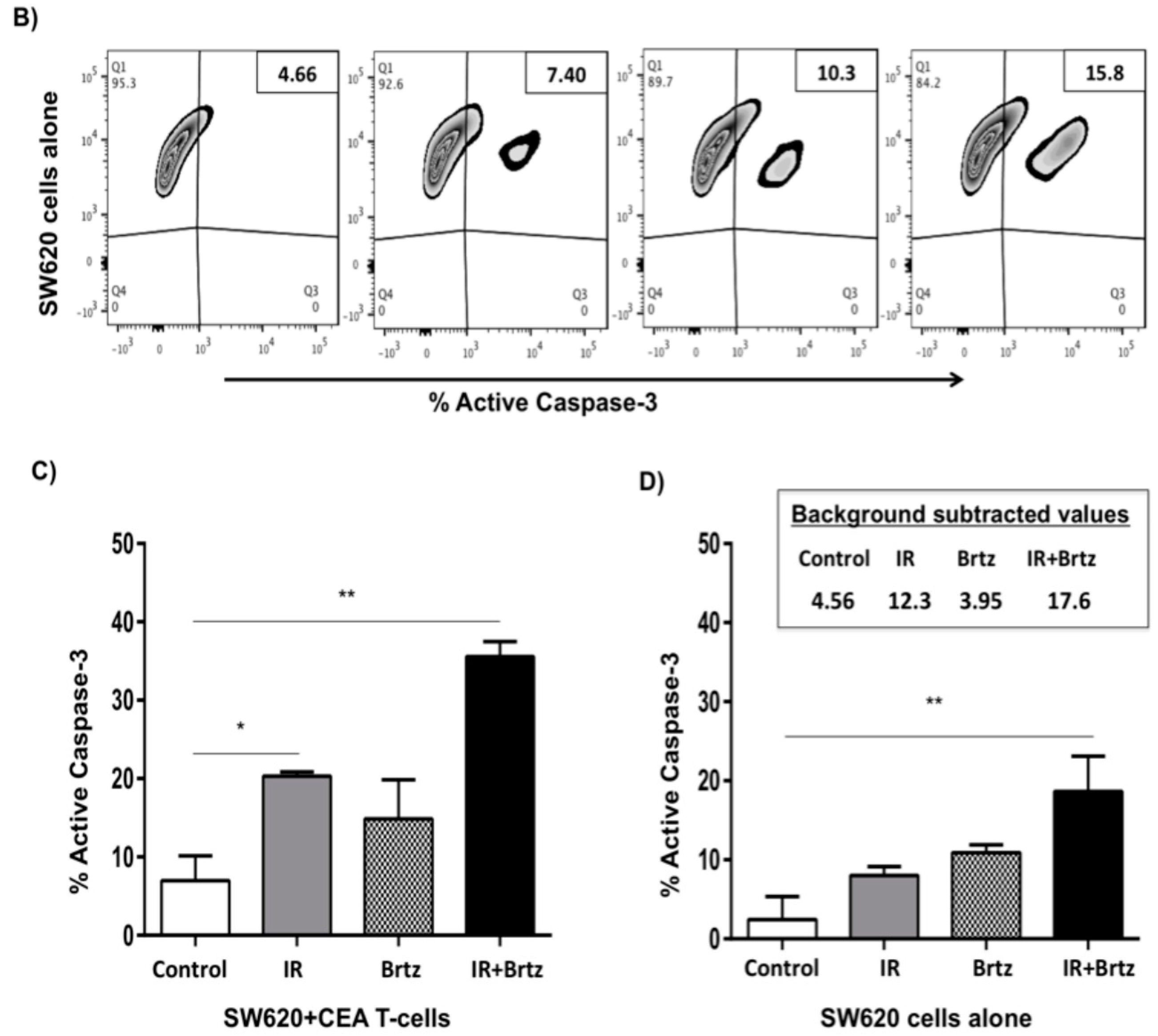
2.5. Proteasome Inhibition Can Further Increase Radiation-Induced Sensitivity to Killing of CRC Cells by CD8+ T Cells



2.6. Proteasome Inhibition Can Further Increase Radiation-Induced Sensitivity to Killing through FasL and TRAIL Receptors

3. Discussion
4. Experimental Section
4.1. Reagents and Cell Lines
4.2. Irradiation
4.3. Apoptosis Assay
4.4. Comet Assay
4.5. RNA Expression and Quantitative Real-Time PCR
4.6. Cell Surface Staining and Flow Cytometry Analysis
4.7. CTL Killing Assay
4.8. Functional Death Receptor Assay
4.9. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. J. Cancer Res. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Akita, H.; Takeda, T.; Nakamura, K.; Kobayashi, S.; Takeda, H. Immunotherapy and hyperthermia for the treatment of patients with advanced or recurrent colorectal cancer. Gan to kagaku ryoho. Cancer Chemother. 2013, 40, 1606–1608. [Google Scholar]
- Ahmed, M.M.; Guha, C.; Hodge, J.W.; Jaffee, E. Immunobiology of radiotherapy: New paradigms. Radiat. Res. 2014, 182, 123–125. [Google Scholar] [CrossRef] [PubMed]
- Casati, A.; Varghaei-Nahvi, A.; Feldman, S.A.; Assenmacher, M.; Rosenberg, S.A.; Dudley, M.E.; Scheffold, A. Clinical-scale selection and viral transduction of human naive and central memory CD8+ T cells for adoptive cell therapy of cancer patients. Cancer Immunol. Immunother. 2013, 62, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Chen, Y.; Xiao, W.; Sun, R.; Tian, Z. NK cell-based immunotherapy for malignant diseases. Cell. Mol. Immunol. 2013, 10, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Grimm, M.; Kim, M.; Rosenwald, A.; von Raden, B.H.; Tsaur, I.; Meier, E.; Heemann, U.; Germer, C.T.; Gasser, M.; Waaga-Gasser, A.M. Tumour-mediated TRAIL-Receptor expression indicates effective apoptotic depletion of infiltrating CD8+ immune cells in clinical colorectal cancer. Eur. J. Cancer 2010, 46, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Pryczynicz, A.; Guzinska-Ustymowicz, K.; Kemona, A. Fas/FasL expression in colorectal cancer. An immunohistochemical study. Folia Histochem. Cytobiol. 2010, 48, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Liu, J.Y.; Xu, H.W.; Yang, C.M.; Zhang, A.Z.; Cui, Y.; Wang, H.B. Mechanism of counterattack of colorectal cancer cell by Fas/Fas ligand system. World J. Gastroenterol. 2005, 11, 6125–6129. [Google Scholar] [CrossRef] [PubMed]
- Petak, I.; Danam, R.P.; Tillman, D.M.; Vernes, R.; Howell, S.R.; Berczi, L.; Kopper, L.; Brent, T.P.; Houghton, J.A. Hypermethylation of the gene promoter and enhancer region can regulate Fas expression and sensitivity in colon carcinoma. Cell Death Differ. 2003, 10, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Koornstra, J.J.; Kleibeuker, J.H.; van Geelen, C.M.; Rijcken, F.E.; Hollema, H.; de Vries, E.G.; de Jong, S. Expression of TRAIL (TNF-related apoptosis-inducing ligand) and its receptors in normal colonic mucosa, adenomas, and carcinomas. J. Pathol. 2003, 200, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Mirandola, P.; Ponti, C.; Gobbi, G.; Sponzilli, I.; Vaccarezza, M.; Cocco, L.; Zauli, G.; Secchiero, P.; Manzoli, F.A.; Vitale, M. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 2004, 104, 2418–2424. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.E.; El-Deiry, W.S. Regulation of the human TRAIL gene. Cancer Biol. Ther. 2012, 13, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Kykalos, S.; Mathaiou, S.; Karayiannakis, A.J.; Patsouras, D.; Lambropoulou, M.; Simopoulos, C. Tissue expression of the proteins fas and fas ligand in colorectal cancer and liver metastases. J. Gastrointest. Cancer 2012, 43, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Perraud, A.; Akil, H.; Nouaille, M.; Petit, D.; Labrousse, F.; Jauberteau, M.O.; Mathonnet, M. Expression of p53 and DR5 in normal and malignant tissues of colorectal cancer: Correlation with advanced stages. Oncol. Rep. 2011, 26, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Ifeadi, V.; Garnett-Benson, C. Sub-lethal irradiation of human colorectal tumor cells imparts enhanced and sustained susceptibility to multiple death receptor signaling pathways. PLoS ONE 2012, 7, e31762. [Google Scholar]
- Kumari, A.; Cacan, E.; Greer, S.F.; Garnett-Benson, C. Turning T cells on: Epigenetically enhanced expression of effector T-cell costimulatory molecules on irradiated human tumor cells. J. Immunother. Cancer 2013, 1, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Garnett, C.T.; Palena, C.; Chakraborty, M.; Tsang, K.Y.; Schlom, J.; Hodge, J.W. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004, 64, 7985–7994. [Google Scholar] [CrossRef] [PubMed]
- Agassi, A.M.; Myslicki, F.A.; Shulman, J.M.; Rotterman, Y.; Dosoretz, D.E.; Fernandez, E.; Mantz, C.A.; Finkelstein, S.E. The promise of combining radiation therapy and immunotherapy: Morbidity and toxicity. Future Oncol. 2014, 10, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dou, Q.P. The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr. Protein Pept. Sci. 2010, 11, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Kinyamu, H.K.; Jefferson, W.N.; Archer, T.K. Intersection of nuclear receptors and the proteasome on the epigenetic landscape. Environ. Mol. Mutagen. 2008, 49, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.; Greer, S.F. Proteolytic and non-proteolytic roles of ubiquitin and the ubiquitin proteasome system in transcriptional regulation. Biochim. Biophys. Acta 2011, 1809, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Bross, P.F.; Kane, R.; Farrell, A.T.; Abraham, S.; Benson, K.; Brower, M.E.; Bradley, S.; Gobburu, J.V.; Goheer, A.; Lee, S.L.; et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 2004, 10, 3954–3964. [Google Scholar] [CrossRef] [PubMed]
- Niewerth, D.; Dingjan, I.; Cloos, J.; Jansen, G.; Kaspers, G. Proteasome inhibitors in acute leukemia. Expert Rev. Anticancer Ther. 2013, 13, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, L.Y.; Vo, D.D.; Garban, H.J.; Comin-Anduix, B.; Owens, S.K.; Dissette, V.B.; Glaspy, J.A.; McBride, W.H.; Bonavida, B.; Economou, J.S.; et al. Immunosensitization of tumor cells to dendritic cell-activated immune responses with the proteasome inhibitor bortezomib (PS-341, Velcade). J. Immunol. 2006, 176, 4757–4765. [Google Scholar] [CrossRef] [PubMed]
- Shanker, A.; Brooks, A.D.; Tristan, C.A.; Wine, J.W.; Elliott, P.J.; Yagita, H.; Takeda, K.; Smyth, M.J.; Murphy, W.J.; Sayers, T.J. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J. Natl. Cancer Inst. 2008, 100, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Seki, N.; Toh, U.; Sayers, T.J.; Fujii, T.; Miyagi, M.; Akagi, Y.; Kusukawa, J.; Kage, M.; Shirouzu, K.; Yamana, H. Bortezomib sensitizes human esophageal squamous cell carcinoma cells to TRAIL-mediated apoptosis via activation of both extrinsic and intrinsic apoptosis pathways. Mol. Cancer Ther. 2010, 9, 1842–1851. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Foster, N.R.; Meyers, J.P.; Thomas, S.P.; Northfelt, D.W.; Rowland, K.M., Jr.; Mattar, B.I.; Johnson, D.B.; Molina, J.R.; Mandrekar, S.J.; et al. A Phase I/II Study of Bortezomib in Combination with Paclitaxel, Carboplatin and Concurrent Thoracic Radiation Therapy for Non-Small Cell Lung Cancer: NCCTG-N0321. J. Thorac. Oncol. 2014, 10, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Peng, S.; Knoff, J.; Lee, S.; Yang, B.; Wu, T.C.; Hung, C.F. Combination of proteasome and HDAC inhibitor enhances HPV16 E7-specific CD8+ T cell immune response and antitumor effects in a preclinical cervical cancer model. J. Biomed. Sci. 2015, 22, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Ostling, O.; Johanson, K.J. Microelectrophoretic study of radiation-induced DNA damages in individual mammalian cells. Biochem. Biophys. Res. Commun. 1984, 123, 291–298. [Google Scholar] [CrossRef]
- Calini, V.; Urani, C.; Camatini, M. Comet assay evaluation of DNA single- and double-strand breaks induction and repair in C3H10T1/2 cells. Cell Biol. Toxicol. 2002, 18, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Huerta, S.; Heinzerling, J.H.; Anguiano-Hernandez, Y.M.; Huerta-Yepez, S.; Lin, J.; Chen, D.; Bonavida, B.; Livingston, E.H. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: Roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/DIABLO, and AIF. J. Surg. Res. 2007, 142, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Jove, V.; Chang, S.; Hedvat, M.; Liu, L.; Buettner, R.; Tian, Y.; Scuto, A.; Wen, W.; Yip, M.L.; et al. Bortezomib induces apoptosis and growth suppression in human medulloblastoma cells, associated with inhibition of AKT and NF-kB signaling, and synergizes with an ERK inhibitor. Cancer Biol. Ther. 2012, 13, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Jane, E.P.; Premkumar, D.R.; Pollack, I.F. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-kB signaling pathway. Mol. Cancer Ther. 2011, 10, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Sunwoo, J.B.; Chen, Z.; Dong, G.; Yeh, N.; Crowl Bancroft, C.; Sausville, E.; Adams, J.; Elliott, P.; van Waes, C. Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 2001, 7, 1419–1428. [Google Scholar] [PubMed]
- Wright, J.J. Combination therapy of bortezomib with novel targeted agents: An emerging treatment strategy. Clin. Cancer Res. 2010, 16, 4094–4104. [Google Scholar] [CrossRef] [PubMed]
- Uziel, O.; Cohen, O.; Beery, E.; Nordenberg, J.; Lahav, M. The effect of Bortezomib and Rapamycin on Telomerase Activity in Mantle Cell Lymphoma. Transl. Oncol. 2014, 7, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Huang, T.C.; Chen, N.N.; Huang, H.C.; Juan, H.F. Combination therapy targeting ectopic ATP synthase and 26S proteasome induces ER stress in breast cancer cells. Cell Death Dis. 2014, 5, e1540. [Google Scholar] [CrossRef] [PubMed]
- Kunami, N.; Katsuya, H.; Nogami, R.; Ishitsuka, K.; Tamura, K. Promise of combining a Bcl-2 family inhibitor with bortezomib or SAHA for adult T-cell leukemia/lymphoma. Anticancer Res. 2014, 34, 5287–5294. [Google Scholar] [PubMed]
- Pervan, M.; Iwamoto, K.S.; McBride, W.H. Proteasome structures affected by ionizing radiation. Mol. Cancer Res. 2005, 3, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The immunobiology of cancer immunosurveillance and immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Blattman, J.; Greenberg, P. Cancer immunotherapy: A treatment for the masses. Science 2004, 305, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Casiano, C.A.; Peng, X.X.; Koziol, J.A.; Chan, E.K.; Tan, E.M. Enhancement of antibody detection in cancer using panel of recombinant tumor-associated antigens. Cancer Epidemiol. Biomark. Prev. 2003, 12, 136–143. [Google Scholar]
- Tsang, K.Y.; Zaremba, S.; Nieroda, C.A.; Zhu, M.Z.; Hamilton, J.M.; Schlom, J. Generation of human cytotoxic T cells specific for human carcinoembryonic antigen epitopes from patients immunized with recombinant vaccinia-CEA vaccine. J. Natl. Cancer Inst. 1995, 87, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Gulley, J.L.; Arlen, P.M.; Tsang, K.Y.; Yokokawa, J.; Palena, C.; Poole, D.J.; Remondo, C.; Cereda, V.; Jones, J.L.; Pazdur, M.P.; et al. Pilot study of vaccination with recombinant CEA-MUC-1-TRICOM poxviral-based vaccines in patients with metastatic carcinoma. Clin. Cancer Res. 2008, 14, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Lesterhuis, W.J.; de Vries, I.J.; Schreibelt, G.; Schuurhuis, D.H.; Aarntzen, E.H.; de Boer, A.; Scharenborg, N.M.; van De Rakt, M.; Hesselink, E.J.; Figdor, C.G.; et al. Immunogenicity of dendritic cells pulsed with CEA peptide or transfected with CEA mRNA for vaccination of colorectal cancer patients. Anticancer Res. 2010, 30, 5091–5097. [Google Scholar] [PubMed]
- Mohebtash, M.; Tsang, K.Y.; Madan, R.A.; Huen, N.Y.; Poole, D.J.; Jochems, C.; Jones, J.; Ferrara, T.; Heery, C.R.; Arlen, P.M.; et al. A pilot study of MUC-1/CEA/TRICOM poxviral-based vaccine in patients with metastatic breast and ovarian cancer. Clin. Cancer Res. 2011, 17, 7164–7173. [Google Scholar] [CrossRef] [PubMed]
- Tawa, P.; Tam, J.; Cassady, R.; Nicholson, D.W.; Xanthoudakis, S. Quantitative analysis of fluorescent caspase substrate cleavage in intact cells and identification of novel inhibitors of apoptosis. Cell Death Differ. 2001, 8, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Andtbacka, R.H.; Dunner, K., Jr.; Pal, A.; Bornmann, W.G.; Chiao, P.J.; Huang, P.; et al. Aggresome disruption: A novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006, 66, 3773–3781. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, M.D.; Pebernard, S.; Iggo, R.D. Regulation of p53 stability and function in HCT116 colon cancer cells. J. Biol. Chem. 2004, 279, 7598–7605. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, N.R.; Rowan, A.; Smith, M.E.; Kerr, I.B.; Bodmer, W.F.; Gannon, J.V.; Lane, D.P. p53 mutations in colorectal cancer, Proc. Natl. Acad. Sci. USA 1990, 87, 7555–7559. [Google Scholar] [CrossRef]
- Mariadason, J.M.; Arango, D.; Shi, Q.; Wilson, A.J.; Corner, G.A.; Nicholas, C.; Aranes, M.J.; Lesser, M.; Schwartz, E.L.; Augenlicht, L.H. Gene expression profiling-based prediction of response of colon carcinoma cells to 5-fluorouracil and camptothecin. Cancer Res. 2003, 63, 8791–8812. [Google Scholar] [PubMed]
- Finkelstein, S.E.; Fishman, M. Clinical opportunities in combining immunotherapy with radiation therapy. Front. Oncol. 2012, 2, 169. [Google Scholar] [CrossRef] [PubMed]
- Weber, D.C.; Ares, C.; Lomax, A.J.; Kurtz, J.M. Radiation therapy planning with photons and protons for early and advanced breast cancer: An overview. Radiat. Oncol. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E.; Ali, M.W.; Boyd, N.H.; Hooks, S.B.; Greer, S.F. Inhibition of HDAC1 and DNMT1 modulate RGS10 expression and decrease ovarian cancer chemoresistance. PLoS ONE 2014, 9, e87455. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cacan, E.; Spring, A.M.; Kumari, A.; Greer, S.F.; Garnett-Benson, C. Combination Treatment with Sublethal Ionizing Radiation and the Proteasome Inhibitor, Bortezomib, Enhances Death-Receptor Mediated Apoptosis and Anti-Tumor Immune Attack. Int. J. Mol. Sci. 2015, 16, 30405-30421. https://doi.org/10.3390/ijms161226238
Cacan E, Spring AM, Kumari A, Greer SF, Garnett-Benson C. Combination Treatment with Sublethal Ionizing Radiation and the Proteasome Inhibitor, Bortezomib, Enhances Death-Receptor Mediated Apoptosis and Anti-Tumor Immune Attack. International Journal of Molecular Sciences. 2015; 16(12):30405-30421. https://doi.org/10.3390/ijms161226238
Chicago/Turabian StyleCacan, Ercan, Alexander M. Spring, Anita Kumari, Susanna F. Greer, and Charlie Garnett-Benson. 2015. "Combination Treatment with Sublethal Ionizing Radiation and the Proteasome Inhibitor, Bortezomib, Enhances Death-Receptor Mediated Apoptosis and Anti-Tumor Immune Attack" International Journal of Molecular Sciences 16, no. 12: 30405-30421. https://doi.org/10.3390/ijms161226238
APA StyleCacan, E., Spring, A. M., Kumari, A., Greer, S. F., & Garnett-Benson, C. (2015). Combination Treatment with Sublethal Ionizing Radiation and the Proteasome Inhibitor, Bortezomib, Enhances Death-Receptor Mediated Apoptosis and Anti-Tumor Immune Attack. International Journal of Molecular Sciences, 16(12), 30405-30421. https://doi.org/10.3390/ijms161226238