
Reconciling Experiment and Theory in the Use of Aryl-Extended Calix[4]pyrrole Receptors for the Experimental Quantification of Chloride–π Interactions in Solution





Abstract
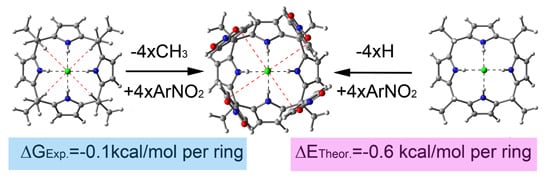
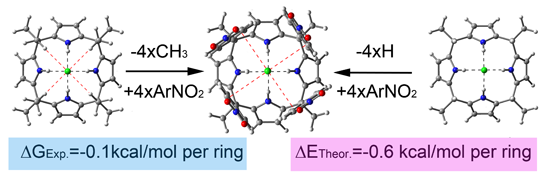
:
1. Introduction

2. Results and Discussion
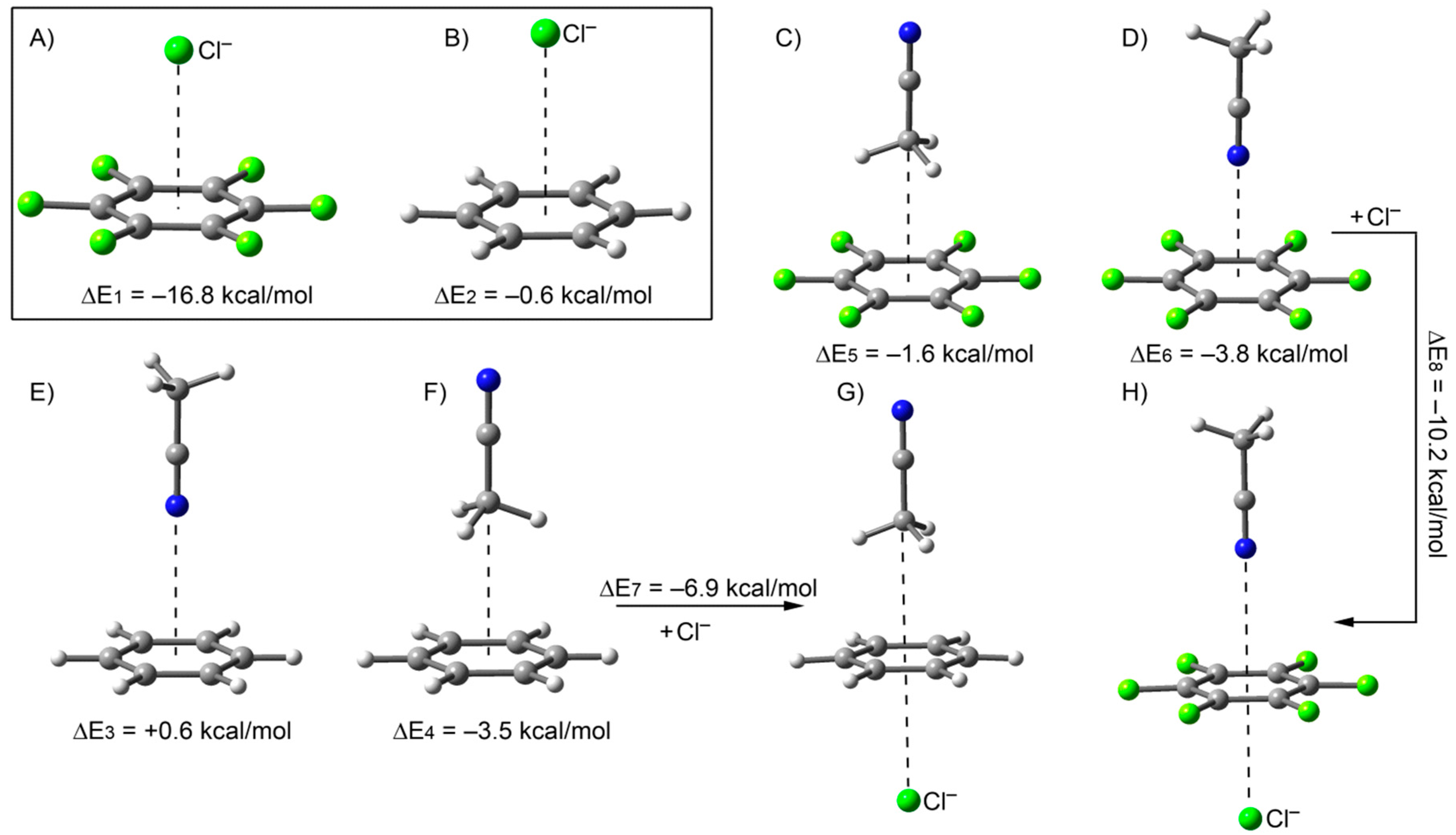
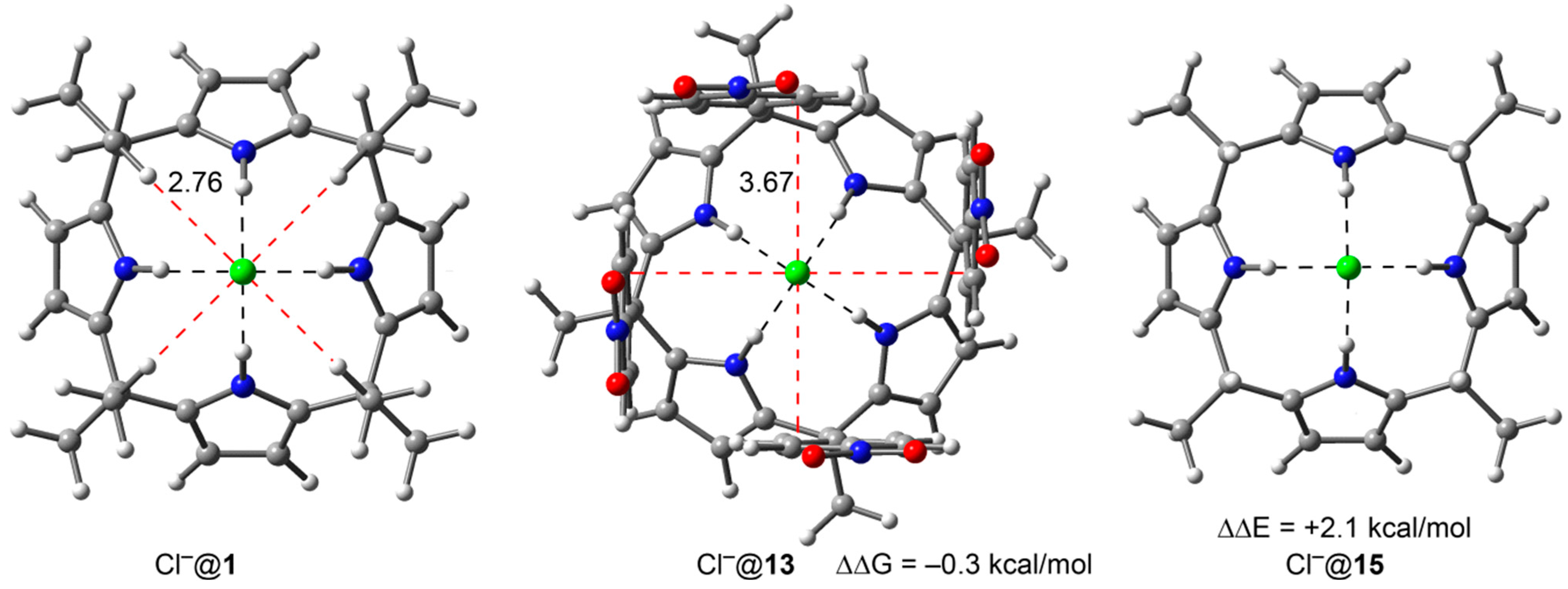
2.1. Solvation Effects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
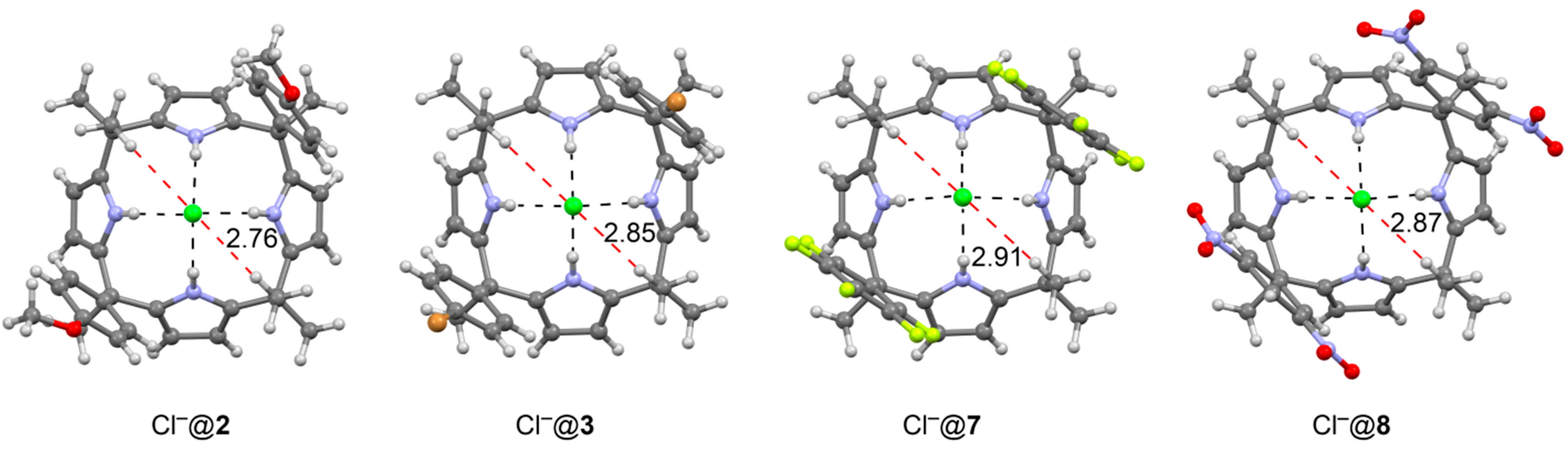{kind=link}
{kind=link}
{kind=link}
| Compound | Aryl | Walls | −ΔGexp | ΔΔGexp | ΔΔEtheor | ΔΔGexp (SC) | ΔΔEtheor (SC) |
|---|---|---|---|---|---|---|---|
| Cl−@1 | none | none | 6.9 | – | – | – | – |
| Cl−@2 | R = OMe, R1 = H | two | 5.5 | 1.4 | 4.5 | 0.7 | 2.3 |
| Cl−@3 | R = Br, R1 = H | two | 6.6 | 0.3 | 0.8 | 0.1 | 0.4 |
| Cl−@4 | R = R1 = H | two | 6.0 | 0.9 | 1.6 | 0.4 | 0.8 |
| Cl−@5 | R = N3 | two | 6.5 | 0.4 | −1.7 | 0.2 | −0.8 |
| Cl−@6 | R = NO2 | two | 7.4 | −0.5 | −5.4 | −0.2 | −2.7 |
| Cl−@7 | C6F5 | two | 7.8 | −0.9 | −7.4 | −0.4 | −3.7 |
| Cl−@8 | R = H, R1 = NO2 | two | 8.5 | −1.6 | −12.1 | −0.8 | −6.0 |
| Cl−@9 | R = OMe | four | 2.9 | 4.0 | 10.5 | 1.0 | 2.6 |
| Cl−@10 | R = Br | four | 4.8 | 2.1 | – | 0.5 | – |
| Cl−@11 | R = H | four | 3.1 | 3.8 | 8.5 | 0.9 | 2.1 |
| Cl−@12 | R = CN | four | 6.2 | 0.7 | −4.7 | 0.2 | −1.2 |
| Cl−@13 | R = NO2 | four | 7.2 | −0.3 | −3.4 | −0.1 | −0.8 |
| Cl−@14 | R = CH3CO2 | four | 4.1 | 2.8 | 13.0 | 0.7 | 3.2 |

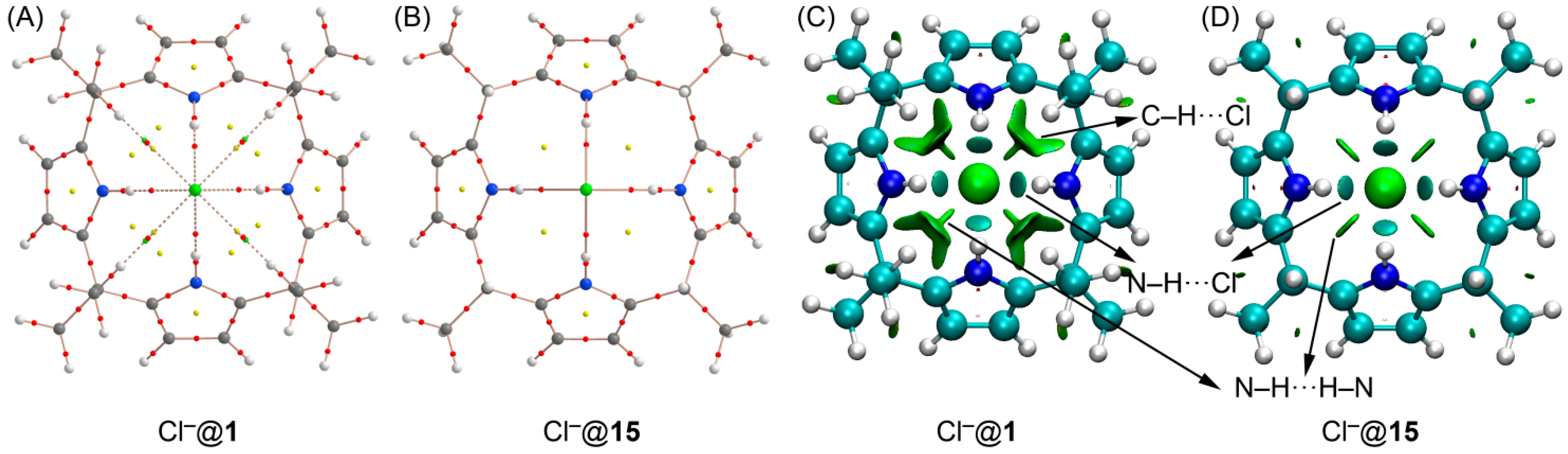
2.2. The Chloride Complex Cl−@1 Used as Reference for Both Model Systems



3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef]
- Schneider, H.J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry; Wiley: Chichester, UK, 2000. [Google Scholar]
- Lehn, J.M. Supramolecular Chemistry Concepts and Perspectives; Wiley-VCH: Weinheim, UK, 1995. [Google Scholar]
- Vögtle, F. Supramolecular Chemistry: An Introduction; Wiley: New York, NY, USA, 1993. [Google Scholar]
- Beer, P.D.; Gale, P.A.; Smith, D.K. Supramolecular Chemistry; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; Wiley: Chichester, UK, 2000. [Google Scholar]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Schneider, H.J.; Yatsimirsky, A. Selectivity in supramolecular host–guest complexes. Chem. Soc. Rev. 2008, 37, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Salonen, L.M.; Ellermann, M.; Diederich, F. Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem. Int. Ed. 2011, 50, 4808–4842. [Google Scholar] [CrossRef]
- Müller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.C.; Dougherty, D.A. The cation–π interaction. Chem. Rev. 1997, 97, 1303–1324. [Google Scholar] [CrossRef] [PubMed]
- Crowley, P.B.; Golovin, A. Cation–π interactions in protein–protein interfaces. Proteins Struct. Funct. Genet. 2005, 59, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Gallivan, J.P.; Dougherty, D.A. A computational study of cation–π interactions vs. salt bridges in aqueous media: Implications for protein engineering. J. Am. Chem. Soc. 2000, 122, 870–874. [Google Scholar] [CrossRef]
- Frontera, A.; Gamez, P.; Mascal, M.; Mooibroek, T.J.; Reedijk, J. Putting anion–π interactions into perspective. Angew. Chem. Int. Ed. 2011, 50, 9564–9583. [Google Scholar] [CrossRef]
- Schottel, B.L.; Chifotides, H.T.; Dunbar, K.R. Anion–π interactions. Chem. Soc. Rev. 2008, 37, 68–83. [Google Scholar] [CrossRef] [PubMed]
- Caltagirone, C.; Gale, P.A. Anion receptor chemistry: Highlights from 2007. Chem. Soc. Rev. 2009, 38, 520–563. [Google Scholar] [CrossRef]
- Gamez, P.; Mooibroek, T.J.; Teat, S.J.; Reedijk, J. Anion binding involving π–acidic heteroaromatic rings. Acc. Chem. Res. 2007, 40, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Hay, B.P.; Bryantsev, V.S. Anion-arene adducts: C–H hydrogen bonding, anion–π interaction, and carbon bonding motifs. Chem. Commun. 2008, 44, 2417–2428. [Google Scholar] [CrossRef]
- Quiñonero, D.; Garau, C.; Rotger, C.; Frontera, A.; Ballester, P.; Costa, A.; Deyà, P.M. Anion–π interactions: Do they exist? Angew. Chem. Int. Ed. 2002, 41, 3389–3392. [Google Scholar] [CrossRef]
- Mascal, M.; Armstrong, A.; Bartberger, M.D. Anion–aromatic bonding: A case for anion recognition by π–acidic rings. J. Am. Chem. Soc. 2002, 124, 6274–6276. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Interaction of anions with perfluoro aromatic compounds. J. Am. Chem. Soc. 2002, 124, 8593–8598. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Singh, N.J.; Kim, K.S. Cyameluric acid as anion–π type receptor for ClO4− and NO3−: π–Stacked and edge-to-face structures. J. Chem. Theor. Comput. 2008, 4, 1401–1407. [Google Scholar] [CrossRef]
- Schottel, B.L.; Bacsa, J.; Dunbar, K.R. Anion dependence of Ag(I) reactions with 3,6-bis(2-pyridyl)-1,2,4,5-tetrazine (bptz): Isolation of the molecular propeller compound [Ag2(bptz)3][AsF6]2. Chem. Commun. 2005, 41, 46–47. [Google Scholar] [CrossRef]
- Han, B.; Lu, J.J.; Kochi, J.K. Anion recognitions via cocrystallizations with organic π–acids in the efficient self-assembly of nanoscopic one-dimensional molecular chains. Cryst. Growth Des. 2008, 8, 1327–1334. [Google Scholar] [CrossRef]
- Mascal, M.; Yakovlev, I.; Nikitin, E.B.; Fettinger, J.C. Fluoride-selective host based on anion–π interactions, ion pairing, and hydrogen bonding: Synthesis and fluoride-ion sandwich complex. Angew. Chem. Int. Ed. 2007, 46, 8782–8784. [Google Scholar] [CrossRef]
- Götz, R.J.; Robertazzi, A.; Mutikainen, I.; Turpeinen, U.; Gamez, P.; Reedijk, J. Concurrent anion–π interactions between a perchlorate ion and two π–acidic aromatic rings, namely pentafluorophenol and 1,3,5-triazine. Chem. Commun. 2008, 44, 3384–3386. [Google Scholar] [CrossRef]
- Albrecht, M.; Müller, M.; Mergel, O.; Rissanen, K.; Valkonen, A. CH-directed anion–π interactions in the crystals of pentafluorobenzyl-substituted ammonium and pyridinium salts. Chem. Eur. J. 2010, 16, 5062–5069. [Google Scholar] [CrossRef] [PubMed]
- Estarellas, C.; Frontera, A.; Quiñonero, D.; Deyà, P.M. Relevant anion–π interactions in biological systems: The case of urate oxidase. Angew. Chem. Int. Ed. 2011, 50, 415–418. [Google Scholar] [CrossRef]
- Chakravarty, S.; Sheng, Z.Z.; Iverson, B.; Moore, B. “η6”-Type anion–π in biomolecular recognition. FEBS Lett. 2012, 586, 4180–4185. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.D.; Harris, J.B.; Howell, E.F.; Hinde, R.J.; Baudry, J. STAAR: STatistical analysis of aromatic rings. J. Comput. Chem. 2013, 34, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, A.V.; Hennig, A.; Mareda, J.; Matile, S. Synthetic ion transporters that work with anion–π interactions, halogen bonds, and anion–macrodipole interactions. Acc. Chem. Res. 2013, 46, 2791–2800. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Beuchat, C.; Domoto, Y.; Gajewy, J.; Wilson, A.; Mareda, J.; Sakai, N.; Matile, S. Anion–π catalysis. J. Am. Chem. Soc. 2014, 136, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Domoto, Y.; Orentas, E.; Beuchat, C.; Emery, D.; Mareda, J.; Sakai, N.; Matile, S. Catalysis with anion–π interactions. Angew. Chem. Int. Ed. 2013, 52, 9940–9943. [Google Scholar] [CrossRef]
- Dawson, R.E.; Hennig, A.; Weimann, D.P.; Emery, D.; Ravikumar, V.; Montenegro, J.; Takeuchi, T.; Gabutti, S.; Mayor, M.; Mareda, J.; et al. Experimental evidence for the functional relevance of anion–π interactions. Nat. Chem. 2010, 2, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Mareda, J.; Vauthey, E.; Matile, S. Core-substituted naphthalenediimides. Chem. Commun. 2010, 46, 4225–4237. [Google Scholar] [CrossRef]
- Gale, P.A.; Caltagirone, C. Anion sensing by small molecules and molecular ensembles. Chem. Soc. Rev. 2015. [Google Scholar] [CrossRef]
- Caballero, A.; Zapata, F.; Beer, P.D. Interlocked host molecules for anion recognition and sensing. Coord. Chem. Rev. 2013, 257, 2434–2455. [Google Scholar] [CrossRef]
- Gale, P.A. Anion receptor chemistry. Chem. Commun. 2011, 47, 82–86. [Google Scholar] [CrossRef]
- Wenzel, M.; Hiscock, J.R.; Gale, P.A. Anion receptor chemistry: Highlights from 2010. Chem. Soc. Rev. 2012, 41, 480–520. [Google Scholar] [CrossRef] [PubMed]
- Gale, P.A.; Quesada, R. Anion coordination and anion-templated assembly: Highlights from 2002 to 2004. Coord. Chem. Rev. 2006, 250, 3219–3244. [Google Scholar] [CrossRef]
- Bowman-James, K.; Bianchi, A.; García-España, E. Anion Coordination Chemistry; Wiley-VCH: Weinheim, UK, 2011. [Google Scholar]
- Berg, J.M. Zinc finger domains: From predictions to design. Acc. Chem. Res. 1995, 28, 14–19. [Google Scholar] [CrossRef]
- Ashcroft, F.M. Ion Channels and Disease; Academic Press: London, UK, 2000. [Google Scholar]
- Moss, B. A land awash with nutrients—The problem of eutrophication. Chem. Ind. 1996, 11, 407–411. [Google Scholar]
- Asakura, T.; Kim, S.Y.; Morita, Y.; Ozawa, M. Reduction of pertechnetate in nitric extraction of rare metals for future reprocessing. J. Nucl. Radiochem. Sci. 2005, 6, 267–269. [Google Scholar] [CrossRef]
- Frontera, A.; Quiñonero, D.; Deyà, P.M. Cation–π and anion–π interactions. WIREs Comput. Mol. Sci. 2011, 1, 440–459. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Krappitz, T.; Peters, M.; Gossen, V.; Raabe, G.; Valkonen, A.; Rissanen, K. Cooperativity of H–bonding and anion–π interaction in the binding of anions with neutral π-acceptors. Chem. Commun. 2012, 48, 9983–9985. [Google Scholar] [CrossRef]
- Ballester, P. Experimental quantification of anion–π interactions in solution using neutral host–guest model systems. Acc. Chem. Res. 2013, 46, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ramírez, G.; Escudero-Adán, E.C.; Benet-Buchholz, J.; Ballester, P. Quantitative evaluation of anion–π interactions in solution. Angew. Chem. Int. Ed. 2008, 47, 4114–4118. [Google Scholar] [CrossRef]
- Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E.C.; Ballester, P. Thermodynamic characterization of halide–π interactions in solution using “two wall” aryl extended calix[4]pyrroles as model system. J. Am. Chem. Soc. 2014, 136, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Blanco, F.; Deyà, P.M.; Elguero, J.; Estarellas, C.; Frontera, A.; Quiñonero, D. Cooperativity in multiple unusual weak bonds. Theor. Chem. Acc. 2010, 126, 1–14. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Elguero, J.; Estarellas, C.; Frontera, A.; Quinonero, D.; Deyà, P.M. Simultaneous interaction of tetrafluoroethene with anions and hydrogen–bond donors: A cooperativity study. J. Chem. Theor. Comput. 2009, 5, 1186–1194. [Google Scholar] [CrossRef]
- Estarellas, C.; Frontera, A.; Quiñonero, D.; Alkorta, I.; Deyà, P.M.; Elguero, J. Energetic vs. synergetic stability: A theoretical study. J. Phys. Chem. A 2009, 113, 3266–3273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhou, B.; Sun, Z.-R.; Wang, X.-B. Photoelectron spectroscopy and theoretical studies of anion–π interactions binding strength and anion specificity. Phys. Chem. Chem. Phys. 2015, 17, 3131–3141. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Hacer, M.; Horn, H.; Kömel, C. Electronic structure calculations on workstation computers: The program system Turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Boys, S.B.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Version 13.05.06; TK Gristmill Software: Overland Park, KS, USA, 2013.
- Johnson, E.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.; Yang, W. Revealing non-covalent interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed]
- Contreras-García, J.; Keinan, S.; Chaudret, R.; Piquemal, J.; Beratan, D.; Yang, W. NCIPLOT: A program for plotting non-covalent interaction regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Contreras-García, J.; Yang, W.; Johnson, E.R. Analysis of hydrogen–bond interaction potentials from the electron density: Integration of non-covalent interaction regions. J. Phys. Chem. A 2011, 115, 12983–12990. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bauzá, A.; Quiñonero, D.; Frontera, A.; Ballester, P. Reconciling Experiment and Theory in the Use of Aryl-Extended Calix[4]pyrrole Receptors for the Experimental Quantification of Chloride–π Interactions in Solution. Int. J. Mol. Sci. 2015, 16, 8934-8948. https://doi.org/10.3390/ijms16048934
Bauzá A, Quiñonero D, Frontera A, Ballester P. Reconciling Experiment and Theory in the Use of Aryl-Extended Calix[4]pyrrole Receptors for the Experimental Quantification of Chloride–π Interactions in Solution. International Journal of Molecular Sciences. 2015; 16(4):8934-8948. https://doi.org/10.3390/ijms16048934
Chicago/Turabian StyleBauzá, Antonio, David Quiñonero, Antonio Frontera, and Pablo Ballester. 2015. "Reconciling Experiment and Theory in the Use of Aryl-Extended Calix[4]pyrrole Receptors for the Experimental Quantification of Chloride–π Interactions in Solution" International Journal of Molecular Sciences 16, no. 4: 8934-8948. https://doi.org/10.3390/ijms16048934
APA StyleBauzá, A., Quiñonero, D., Frontera, A., & Ballester, P. (2015). Reconciling Experiment and Theory in the Use of Aryl-Extended Calix[4]pyrrole Receptors for the Experimental Quantification of Chloride–π Interactions in Solution. International Journal of Molecular Sciences, 16(4), 8934-8948. https://doi.org/10.3390/ijms16048934