
Conformational Ensembles Explored Dynamically from Disordered Peptides Targeting Chemokine Receptor CXCR4






 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Circular Dichroism

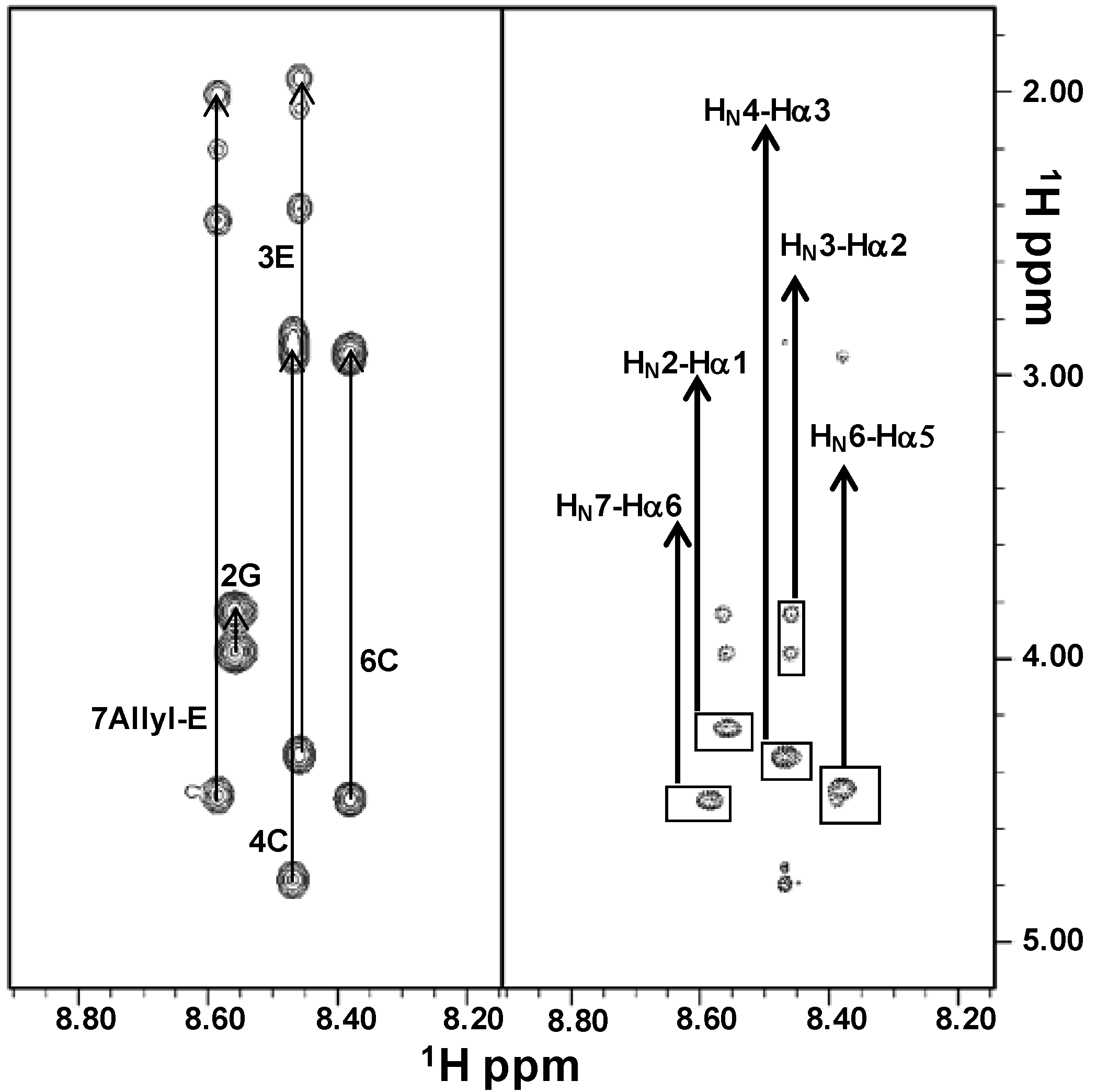
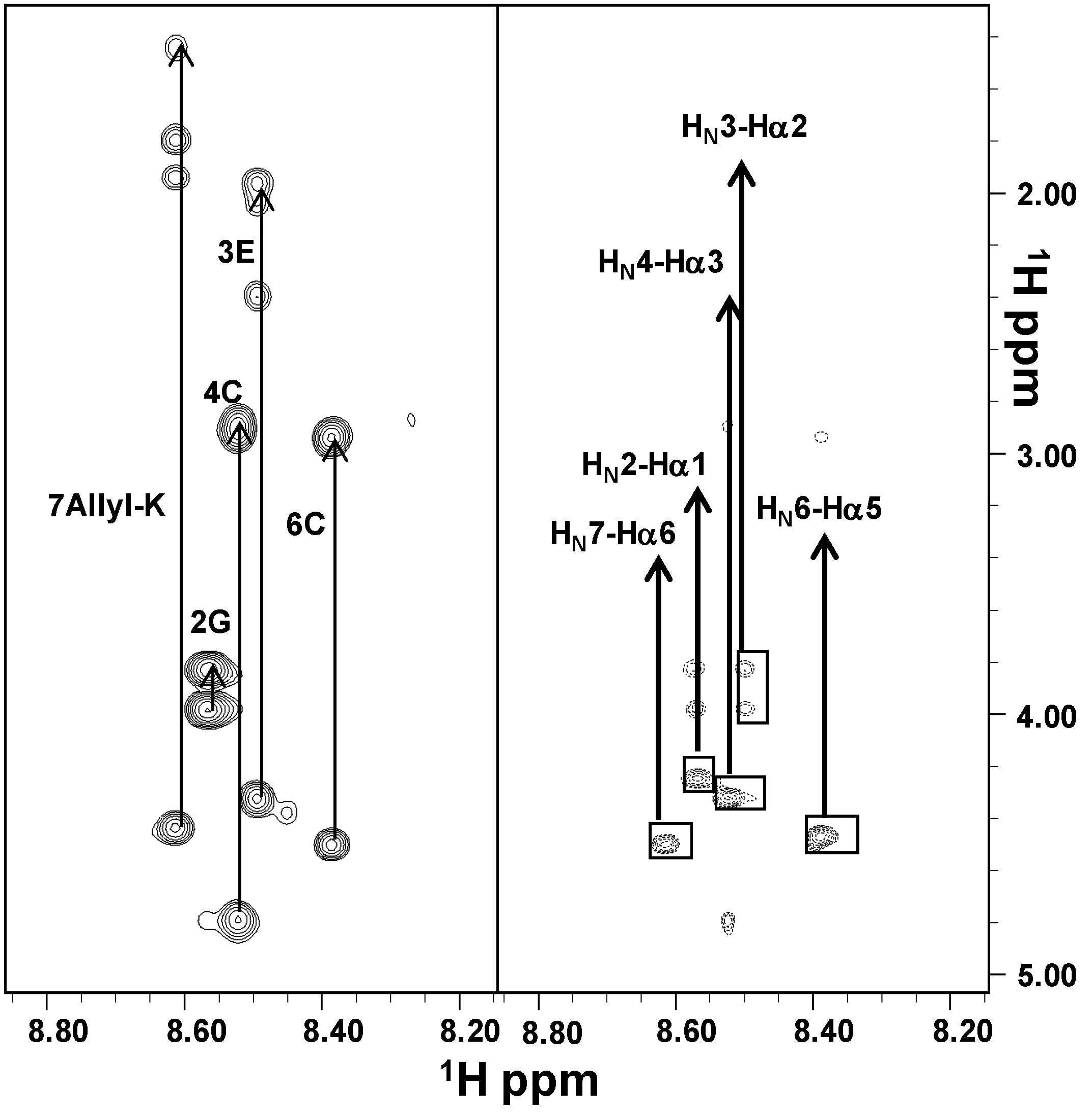
2.2. NMR Characterization


2.3. Molecular Dynamics Simulations on the Two Peptides


2.4. Functional Characterization on CXCR4 Receptor
3. Experimental Section
3.1. Synthesis
3.2. Circular Dichroism Measurement
3.3. NMR Analysis
3.4. Molecular Modeling and Dynamics Simulations
3.5. Migration Assay
3.6. Binding Assay
3.7. cAMP Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar]
- He, B.; Wang, K.; Liu, Y.; Xue, B.; Uversky, V.N.; Dunker, A.K. Predicting intrinsic disorder in proteins: An overview. Cell Res. 2009, 19, 929–949. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, H.N.; Wrabl, J.O.; Li, J.; Hilsen, V.J. The ensemble nature of allostery. Nature 2014, 508, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradovic, Z.; Dunker, A.K. Intrinsicdisorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Schweitzer-Stenner, R. Conformational propensities and residual structures in unfolded peptides and proteins. Mol. BioSyst. 2012, 8, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. Intrinsically disordered protein from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [CrossRef] [PubMed]
- Leone, M.; Mercurio, F.A.; Vincenzi, M.; Accardo, A.; Ringhieri, P.; Tesauro, D.; Carrière, F.; Rossi, F. Conformational disorder in phosphopeptides: Solution studies by CD and NMR techniques. Peptidomics 2014, 1, 14–21. [Google Scholar] [CrossRef]
- Miller, M. The importance of being flexible: The case of basic region leucine zipper transcriptional regulators. Curr. Protein Pept. Sci. 2009, 10, 244–269. [Google Scholar] [CrossRef] [PubMed]
- Sigalov, A.B.; Uversky, V.N. Differential occurrence of protein intrinsic disorder in the cytoplasmic signaling domains of cell receptors. Self Nonself 2011, 2, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Sharma, A.; Raucci, R.; Costantini, M.; Autiero, I.; Colonna, G. Genealogy of an ancient protein family: The Sirtuins, a family of disordered members. BMC Evol. Biol. 2013, 13, 60–80. [Google Scholar] [CrossRef] [PubMed]
- Raucci, R.; Colonna, G.; Giovane, A.; Castello, G.; Costantini, S. N-terminal region of human chemokine receptor CXCR3: Structural analysis of CXCR3(1–48) by experimental and computational studies. Biochim. Biophys. Acta 2014, 1844, 1868–1880. [Google Scholar] [CrossRef] [PubMed]
- Mittal, J.; HyeonYoo, T.; Georgiou, G.; Truskett, T.T. Structural ensemble of an intrinsically disordered polypeptide. J. Phys. Chem. B 2013, 117, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Gao, S.; Cheng, D.; Huang, J. The characterization and comparison of amyloidogenic segments and non-amyloidogenic segments shed light on amyloid formation. Biochem. Biophys. Res. Commun. 2014, 447, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Costantini, S.; Raucci, R.; Colonna, G.; Mercurio, F.A.; Trotta, A.M.; Ringhieri, P.; Leone, M.; Rossi, F.; Pellegrino, C.; Castello, G.; et al. Peptides targeting chemokine receptor CXCR4: Structural behavior and biological binding studies. J. Pept. Sci. 2014, 20, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Palladino, P.; Portella, L.; Colonna, G.; Raucci, R.; Saviano, G.; Rossi, F.; Napolitano, M.; Scala, S.; Castello, G.; Costantini, S. The N-terminal region of CXCL11 as structural template for CXCR3 molecular recognition: Synthesis, conformational analysis, and binding studies. Chem. Biol. Drug Des. 2012, 80, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Underfriend, S.; Meienhofer, J. In The Peptides; Hruby, V.J., Ed.; Academic Press: New York, NY, USA, 1985; Volume 7. [Google Scholar]
- Wiedemann, C.I.; Bellstedt, P.; Görlach, M. CAPITO-a web server-based analysis and plotting tool for circular dichroism data. Bioinformatics 2013, 29, 1750–1757. [Google Scholar] [CrossRef] [PubMed]
- Sreerama, N.; Woody, R.W. Poly(Pro)II helices in globular proteins: Identification and circular dichroic analysis. Biochemistry 1994, 33, 10022–10025. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, R.S.; Gough, C.A. Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA, 1996. [Google Scholar]
- Kumar, A.; Ernst, R.R.; Wuthrich, K. A two-dimensional nuclear Overhauser enhancement (2D NOE) experiment for the elucidation of complete proton-proton cross-relaxation networks in biological macromolecules. Biochem. Biophys. Res. Commun. 1980, 95, 1–6. [Google Scholar] [CrossRef]
- Griesinger, C.; Otting, G.; Wüthrich, K.; Ernst, R.R. Clean TOCSY for proton spin system identification in macromolecules. J. Am. Chem. Soc. 1988, 110, 7870–7872. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. Practical aspects of two-dimensional transverse NOE spectroscopy. J. Magn. Reson. 1985, 63, 207–213. [Google Scholar] [CrossRef]
- Wishart, D.S.; Sykes, B.D.; Richards, F.M. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 1991, 222, 311–333. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, NY, USA, 1986. [Google Scholar]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins areinfluenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed]
- Vila, J.A.; Baldoni, H.A.; Ripoll, D.R.; Ghosh, A.; Scheraga, H.A. PII helix conformation in a proline-rich environment: A theoretical study. Biophys. J. 2004, 86, 731–742. [Google Scholar] [CrossRef]
- Tarek, M.; Tobias, D.J. Role of protein–water hydrogen bond dynamics in the protein dynamical transition. Phys. Rev. Lett. 2002, 88, 138101. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. AMD3100 story: The path to the discovery of a stem cell mobilizer (Mozobil). Biochem. Pharmacol. 2009, 77, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Portella, L.; Vitale, R.; de Luca, S.; D’Alterio, C.; Ieranò, C.; Napolitano, M.; Riccio, A.; Polimeno, M.N.; Monfregola, L.; Barbieri, A.; et al. Preclinical development of a novel class of CXCR4 antagonist impairing solid tumors growth and metastases. PLoS ONE 2013, 8, e74548. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef] [PubMed]
- Piantini, U.; Sorensen, O.W.; Ernst, R.R. Multiple quantum filters for elucidating NMR coupling networks. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar] [CrossRef]
- Dalvit, C. Efficient multiple-solvent suppression for the study of the interactions of organic solvents with biomolecules. J. Biomol. NMR 1998, 11, 437–444. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.H.; Billeter, M.; Guntert, P.; Wuthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Guariniello, S.; Colonna, G.; Raucci, R.; Costantini, M.; di Bernardo, G.; Bergantino, F.; Castello, G.; Costantini, S. Structure-function relationship and evolutionary history of the human selenoprotein M (SelM) found over-expressed in hepatocellular carcinoma. Biochim. Biophys. Acta 2014, 1844, 447–456. [Google Scholar] [CrossRef] [PubMed]
- McDonald, I.K.; Thornton, J.M. Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 1994, 238, 777–793. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincenzi, M.; Costantini, S.; Scala, S.; Tesauro, D.; Accardo, A.; Leone, M.; Colonna, G.; Guillon, J.; Portella, L.; Trotta, A.M.; et al. Conformational Ensembles Explored Dynamically from Disordered Peptides Targeting Chemokine Receptor CXCR4. Int. J. Mol. Sci. 2015, 16, 12159-12173. https://doi.org/10.3390/ijms160612159
Vincenzi M, Costantini S, Scala S, Tesauro D, Accardo A, Leone M, Colonna G, Guillon J, Portella L, Trotta AM, et al. Conformational Ensembles Explored Dynamically from Disordered Peptides Targeting Chemokine Receptor CXCR4. International Journal of Molecular Sciences. 2015; 16(6):12159-12173. https://doi.org/10.3390/ijms160612159
Chicago/Turabian StyleVincenzi, Marian, Susan Costantini, Stefania Scala, Diego Tesauro, Antonella Accardo, Marilisa Leone, Giovanni Colonna, Jean Guillon, Luigi Portella, Anna Maria Trotta, and et al. 2015. "Conformational Ensembles Explored Dynamically from Disordered Peptides Targeting Chemokine Receptor CXCR4" International Journal of Molecular Sciences 16, no. 6: 12159-12173. https://doi.org/10.3390/ijms160612159
APA StyleVincenzi, M., Costantini, S., Scala, S., Tesauro, D., Accardo, A., Leone, M., Colonna, G., Guillon, J., Portella, L., Trotta, A. M., Ronga, L., & Rossi, F. (2015). Conformational Ensembles Explored Dynamically from Disordered Peptides Targeting Chemokine Receptor CXCR4. International Journal of Molecular Sciences, 16(6), 12159-12173. https://doi.org/10.3390/ijms160612159