
A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors

Abstract
:1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | General Structure | Substituents | IC50 (nM) | pIC50 |
|---|---|---|---|---|
| 1 |  | - | 42 | 7.377 |
| 2 b |  | - | 18 | 7.745 |
| 3 |  | - | 247 | 6.607 |
| 4 a |  | H | 61 | 7.215 |
| 5 | Me | 40 | 7.398 | |
| 6 | Et | 59 | 7.229 | |
| 7 a | i-Pr | 60 | 7.222 | |
| 8 | t-Bu | 69 | 7.161 | |
| 9 b | Cyclobutyl | 31 | 7.509 | |
| 10 | 4-Piperidine | 107 | 6.971 | |
| 11 | 3-Piperidine | 167 | 6.777 | |
| 12 |  | H | 3.6 | 8.444 |
| 13 | 4-F | 4.4 | 8.357 | |
| 14 a | 4-Cl | 2.2 | 8.658 | |
| 15 | 4-Br | 2.2 | 8.658 | |
| 16 a | 3-F | 3.1 | 8.509 | |
| 17 | 3-Cl | 1.1 | 8.959 | |
| 18 b | 3-Br | 0.76 | 9.119 | |
| 19 | 2-F | 8.0 | 8.097 | |
| 20 | 2-Cl | 27 | 7.569 | |
| 21 a | 2-Br | 27 | 7.569 | |
| 22 | 3,4-di-F | 3.4 | 8.469 | |
| 23 | 3,4-di-Cl | 1.5 | 8.824 | |
| 24 b | 4-MeO | 1.1 | 8.959 | |
| 25 a | 4-Me | 1.3 | 8.886 | |
| 26 b | 4-CF3 | 1.4 | 8.854 | |
| 27 | 4-CF3O | 2.4 | 8.620 | |
| 28 a | 4-CN | 2.7 | 8.569 | |
| 29 | 4-MeSO2 | 1.4 | 8.854 | |
| 30 | 3-MeO | 1.2 | 8.921 | |
| 31 b | 3-CF3 | 1.0 | 9.000 | |
| 32 a | 4-Pyridyl | 3.2 | 8.495 | |
| 33 | 3-Pyridyl | 3.0 | 8.523 | |
| 34 b |  | H | 1.0 | 9.000 |
| 35 a | 4-F | 1.1 | 8.959 | |
| 36 | 4-Cl | 2.4 | 8.620 | |
| 37 b |  | H | 4.6 | 8.337 |
| 38 | 4-F | 11 | 7.959 | |
| 39 a | 4-Cl | 8.2 | 8.086 |
2. Results and Discussion
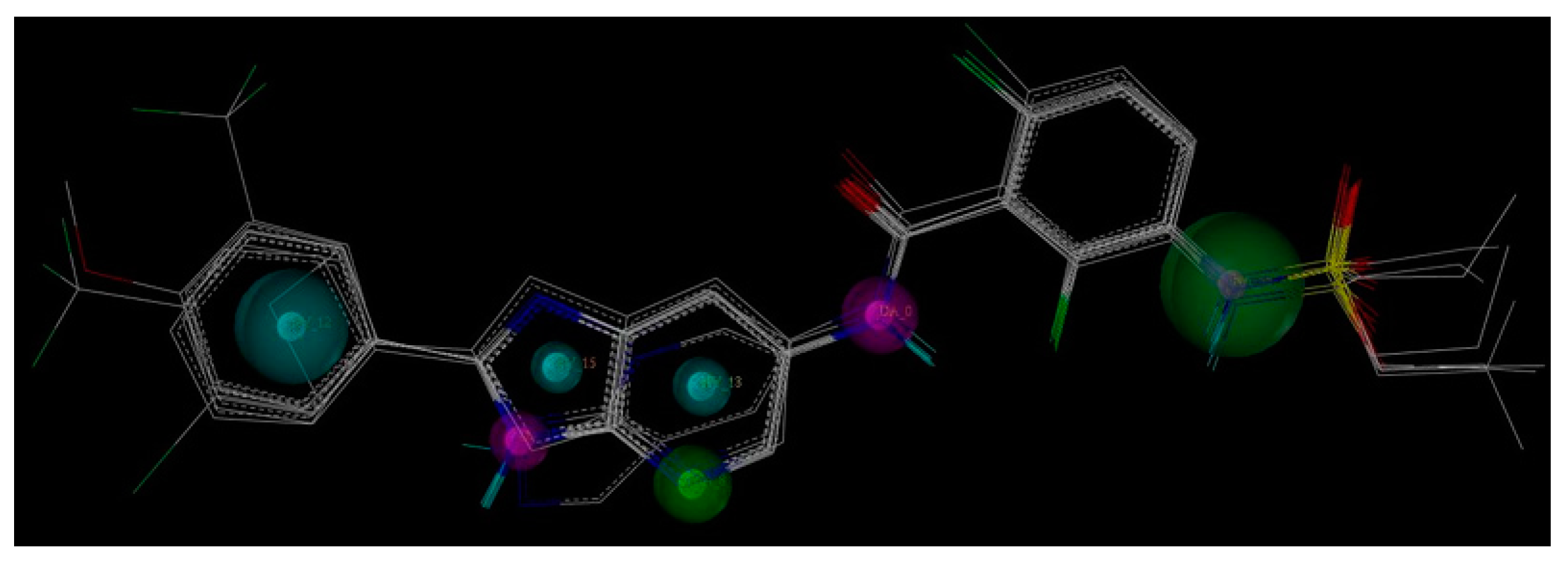
2.1. Pharmacophore Generation
| No. | Specificity | N_hits | Features | Pareto Rank | Energy | Sterics | H-Bond | Mol_Qry |
|---|---|---|---|---|---|---|---|---|
| Model_01 | 4.37 | 8 | 10 | 0 | 11.15 | 3574.20 | 1683.50 | 561.51 |
| Model_02 | 2.93 | 8 | 11 | 0 | 19.35 | 3488.20 | 1822.20 | 381.41 |
| Model_03 | 1.57 | 8 | 13 | 0 | 11.63 | 3390.50 | 1685.10 | 703.76 |
| Model_04 | 1.34 | 8 | 12 | 0 | 18.07 | 3287.20 | 1854.60 | 319.43 |
| Model_05 | 0.24 | 8 | 8 | 0 | 15.77 | 3370.20 | 1791.80 | 282.04 |
| Model_06 | 4.96 | 8 | 8 | 0 | 28.49 | 3242.80 | 1901.20 | 439.41 |
| Model_07 | 3.10 | 8 | 10 | 0 | 19.61 | 3365.90 | 1761.60 | 479.45 |
| Model_08 | −0.15 | 8 | 10 | 0 | 35.19 | 3639.30 | 1767.30 | 367.28 |
| Model_09 | 0.11 | 8 | 8 | 0 | 48.42 | 3312.90 | 1775.00 | 562.73 |
| Model_10 | 2.11 | 8 | 11 | 0 | 25.32 | 3130.60 | 1806.20 | 445.97 |
| Model_11 | 3.53 | 8 | 8 | 0 | 13.04 | 3727.10 | 1786.60 | 165.73 |
| Model_12 | 4.37 | 8 | 10 | 0 | 144.90 | 3504.50 | 1757.90 | 399.20 |
| Model_13 | 3.40 | 8 | 13 | 0 | 10.95 | 2673.80 | 1743.10 | 583.52 |
| Model_14 | 2.48 | 8 | 8 | 0 | 10.03 | 2992.50 | 1768.10 | 249.76 |
| Model_15 | 4.30 | 8 | 10 | 0 | 9.35 | 3189.60 | 1715.70 | 235.40 |
| Model_16 | 2.06 | 8 | 12 | 0 | 13.21 | 2832.40 | 1762.00 | 445.41 |
| Model_17 | 3.12 | 7 | 10 | 0 | 6.94 | 3084.10 | 1587.70 | 304.97 |
| Model_18 | 3.34 | 8 | 9 | 0 | 17.22 | 3054.40 | 1746.00 | 423.70 |
| Model_19 | −0.01 | 8 | 9 | 0 | 13.59 | 3423.70 | 1699.30 | 225.07 |
| Model_20 | 4.82 | 8 | 9 | 0 | 6.82 | 2660.50 | 1576.80 | 355.02 |

2.2. 3D QSAR Studies

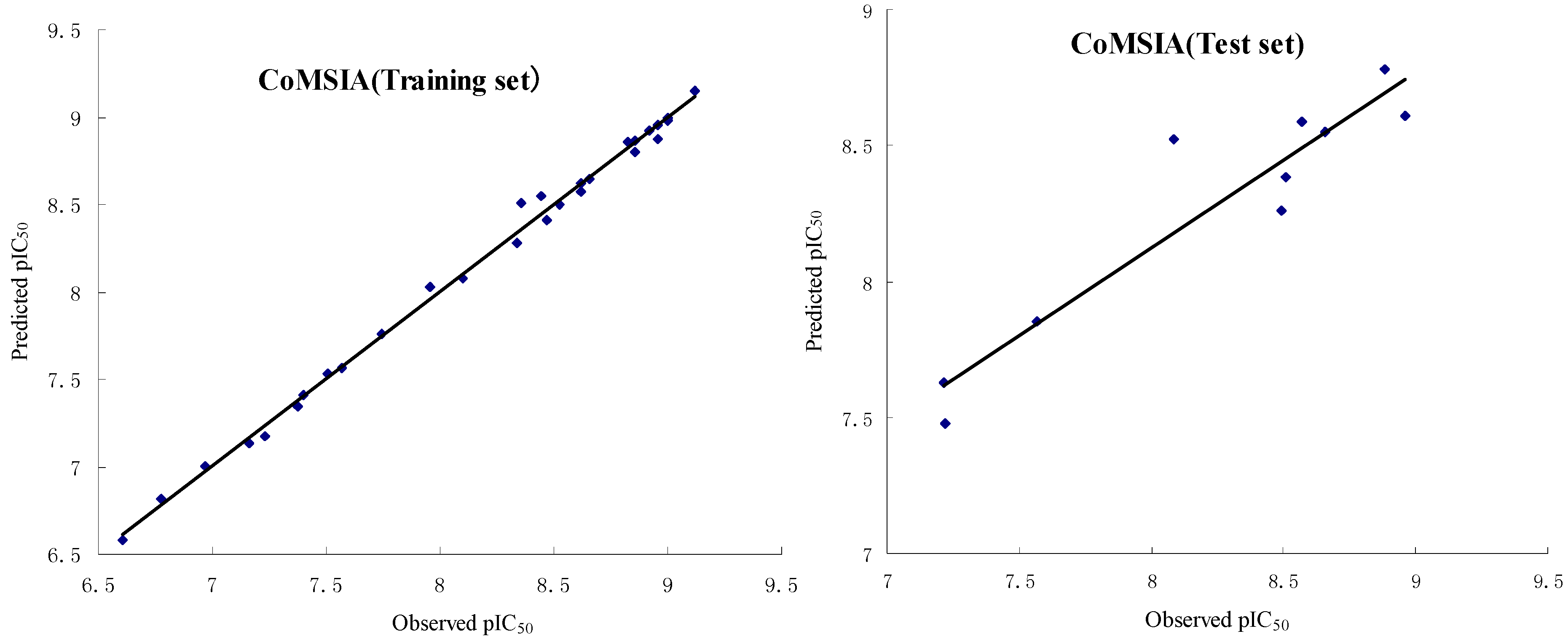
2.2.1. CoMFA and CoMSIA Statistical Results
| Components | Pharmacophore-Based Model | Docking-Based Model | ||
|---|---|---|---|---|
| CoMFA | CoMSIA | CoMFA | CoMSIA | |
| q2(r2cv) | 0.501 | 0.621 | 0.690 | 0.541 |
| SEE | 0.185 | 0.063 | 0.019 | 0.312 |
| F value | 113.846 | 410.567 | 3206.612 | 47.971 |
| r2pred | 0.786 | 0.885 | 0.590 | 0.607 |
| No. of compounds | 29 | 29 | 29 | 29 |
| No. of optimal components | 4 | 10 | 14 | 3 |
| Contributions | ||||
| Steric | 0.579 | 0.196 | 0.542 | 0.185 |
| Electrostatic | 0.421 | 0.201 | 0.458 | 0.185 |
| Hydrophobic | - | 0.291 | 0.338 | |
| H-bond donor | - | 0.161 | 0.165 | |
| H-bond acceptor | - | 0.151 | 0.127 | |
2.2.2. Validation of 3D QSAR Models
| Compound | Observed pIC50 | Pharmacophore-Based CoMSIA | |
|---|---|---|---|
| Predicted pIC50 | Residual | ||
| 1 | 7.377 | 7.343 | 0.034 |
| 2 | 7.745 | 7.761 | −0.016 |
| 3 | 6.607 | 6.583 | 0.024 |
| 4 a | 7.215 | 7.630 | −0.415 |
| 5 | 7.398 | 7.410 | −0.012 |
| 6 | 7.229 | 7.173 | 0.056 |
| 7 a | 7.222 | 7.478 | −0.256 |
| 8 | 7.161 | 7.137 | 0.024 |
| 9 | 7.509 | 7.534 | −0.025 |
| 10 | 6.971 | 7.004 | −0.033 |
| 11 | 6.777 | 6.817 | −0.040 |
| 12 | 8.444 | 8.549 | −0.105 |
| 13 | 8.357 | 8.511 | −0.154 |
| 14 a | 8.658 | 8.551 | 0.107 |
| 15 | 8.658 | 8.645 | 0.013 |
| 16 a | 8.509 | 8.384 | 0.125 |
| 17 | 8.959 | 8.956 | 0.003 |
| 18 | 9.119 | 9.149 | −0.030 |
| 19 | 8.097 | 8.076 | 0.021 |
| 20 | 7.569 | 7.563 | 0.006 |
| 21 a | 7.569 | 7.850 | −0.281 |
| 22 | 8.469 | 8.412 | 0.057 |
| 23 | 8.824 | 8.860 | −0.036 |
| 24 | 8.959 | 8.872 | 0.087 |
| 25 a | 8.886 | 8.782 | 0.104 |
| 26 | 8.854 | 8.802 | 0.052 |
| 27 | 8.620 | 8.572 | 0.048 |
| 28 a | 8.569 | 8.588 | −0.019 |
| 29 | 8.854 | 8.863 | −0.009 |
| 30 | 8.921 | 8.923 | −0.002 |
| 31 | 9.000 | 8.996 | 0.004 |
| 32 a | 8.495 | 8.259 | 0.236 |
| 33 | 8.523 | 8.500 | 0.023 |
| 34 | 9.000 | 8.976 | 0.024 |
| 35 a | 8.959 | 8.608 | 0.351 |
| 36 | 8.620 | 8.618 | 0.002 |
| 37 | 8.337 | 8.283 | 0.054 |
| 38 | 7.959 | 8.030 | −0.071 |
| 39 a | 8.086 | 8.521 | −0.435 |

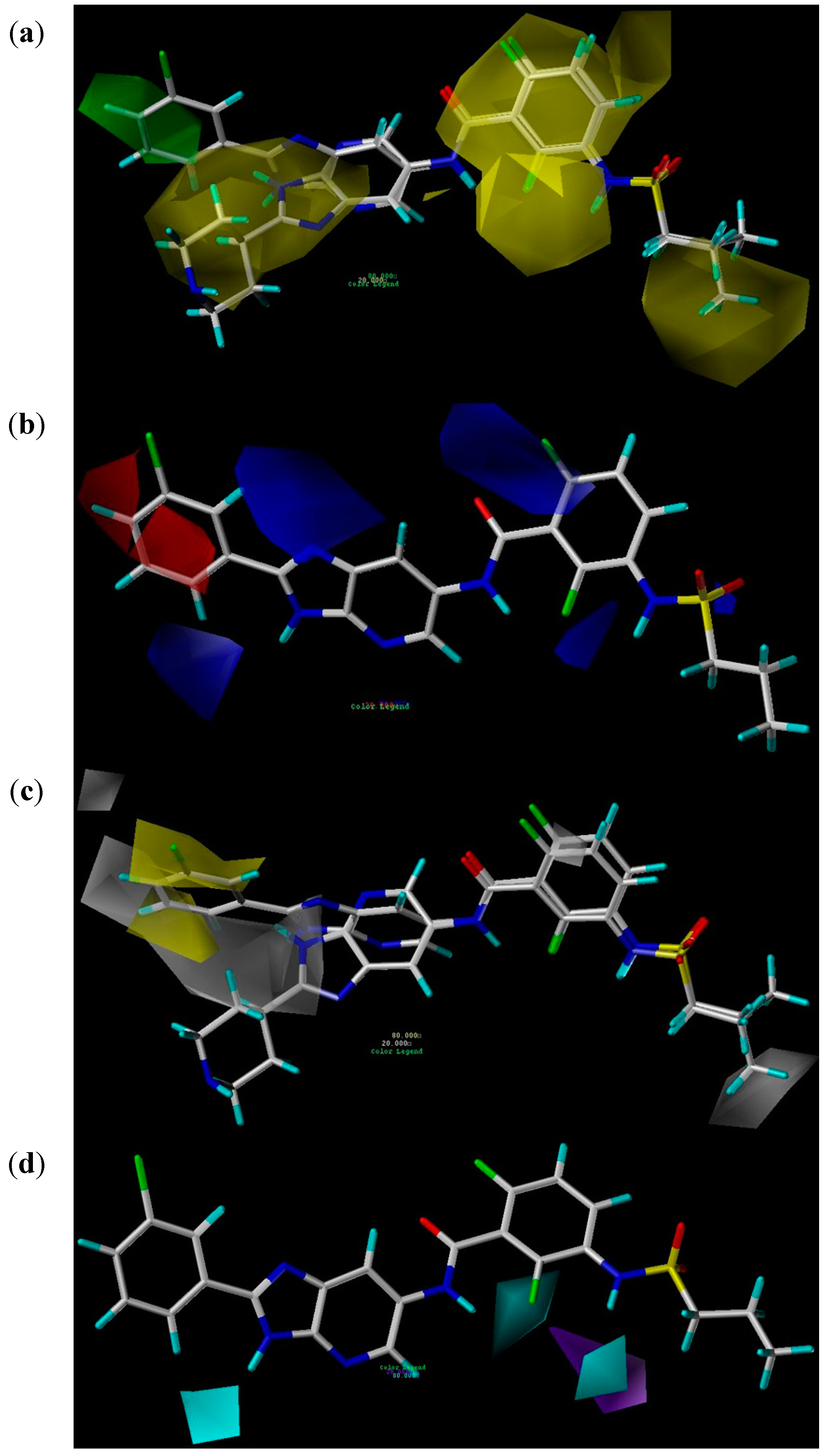
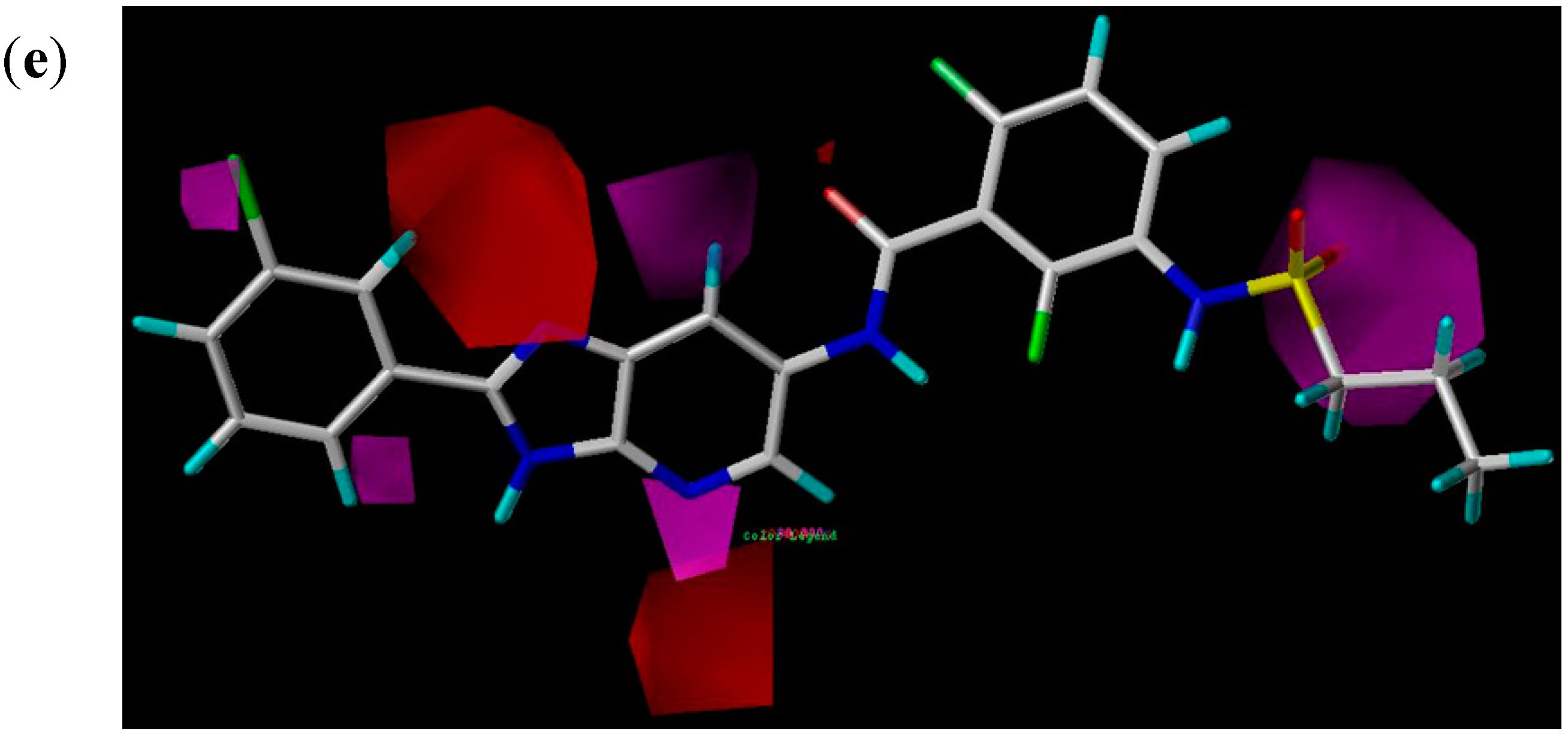
2.2.3. CoMSIA Contour Maps


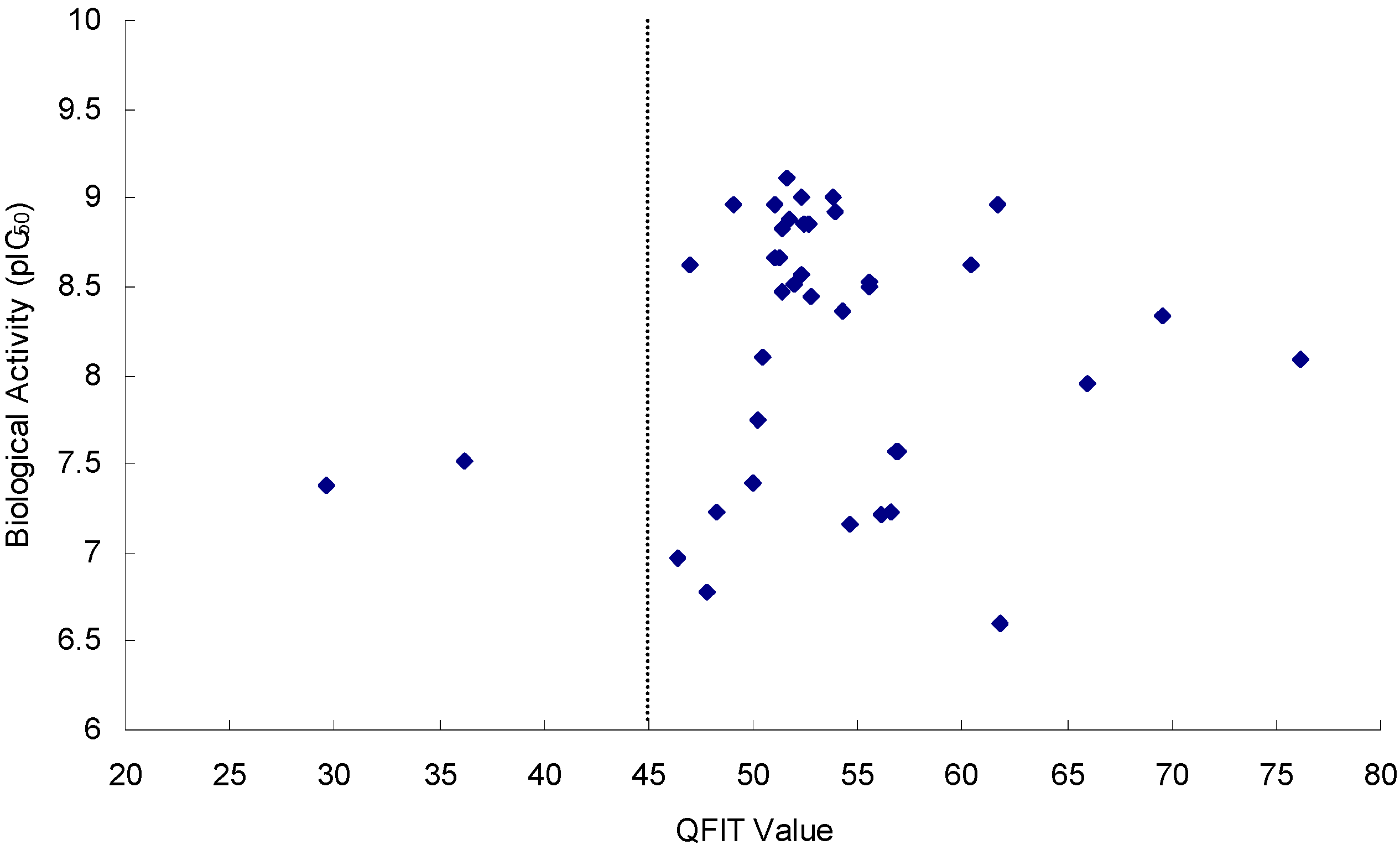
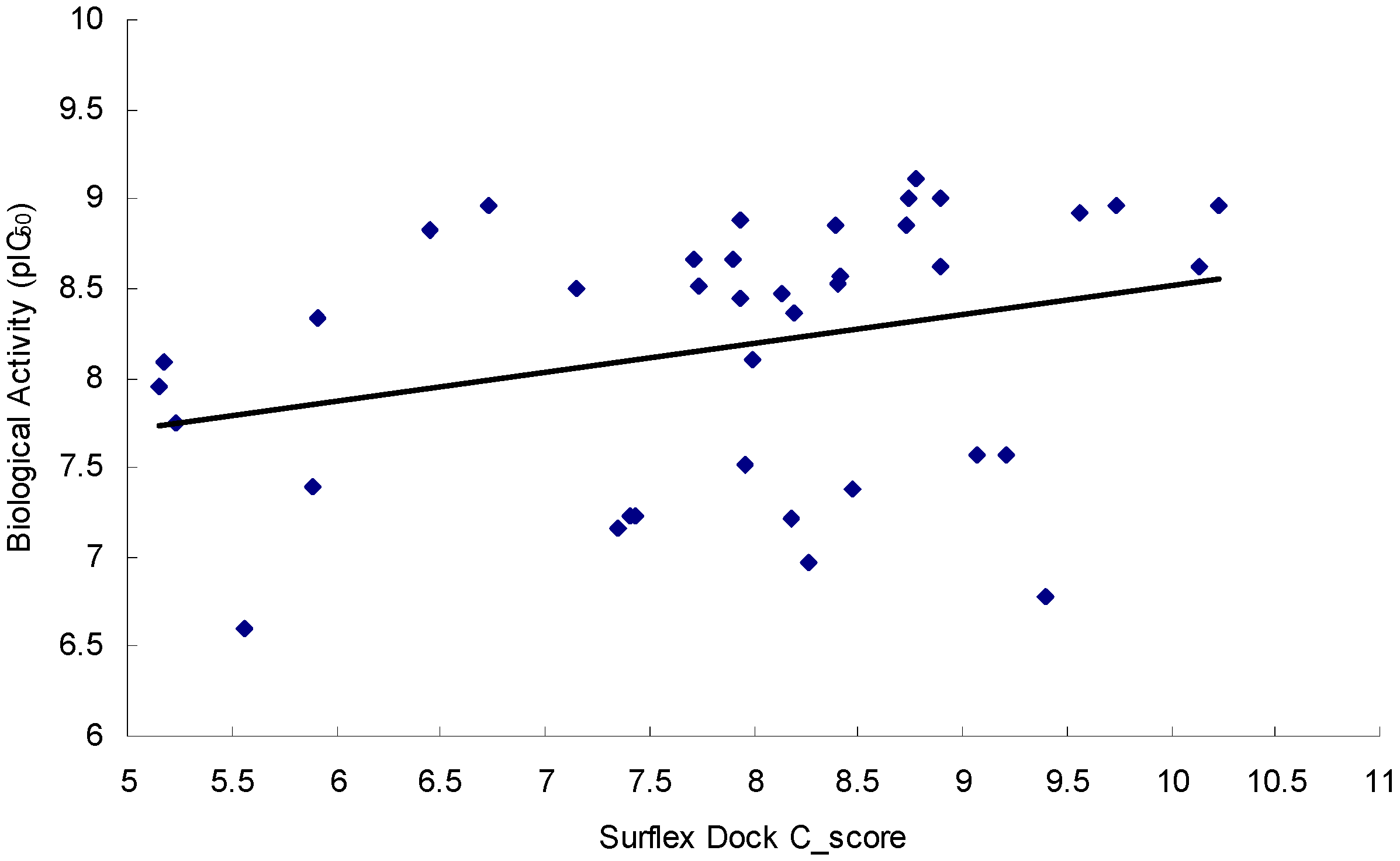
2.3. Virtual Screening
2.3.1. Pharmacophore Model Validation

2.3.2. Docking Model Validation

2.3.3. Screening of NCI2000 Database
| Hit Compound | Structure | QFIT Value | Docking C_Score | Predicted pIC50 |
|---|---|---|---|---|
| NCI 94680 |  | 66.50 | 6.84 | 8.520 |
| NCI 527880 |  | 67.58 | 5.55 | 8.263 |
| NCI 183519 |  | 62.80 | 5.28 | 7.667 |
3. Experimental Section
3.1. Compounds and Biological Data
3.2. Molecular Modeling
3.3. Pharmacophore Hypothesis
3.4. Molecular Docking
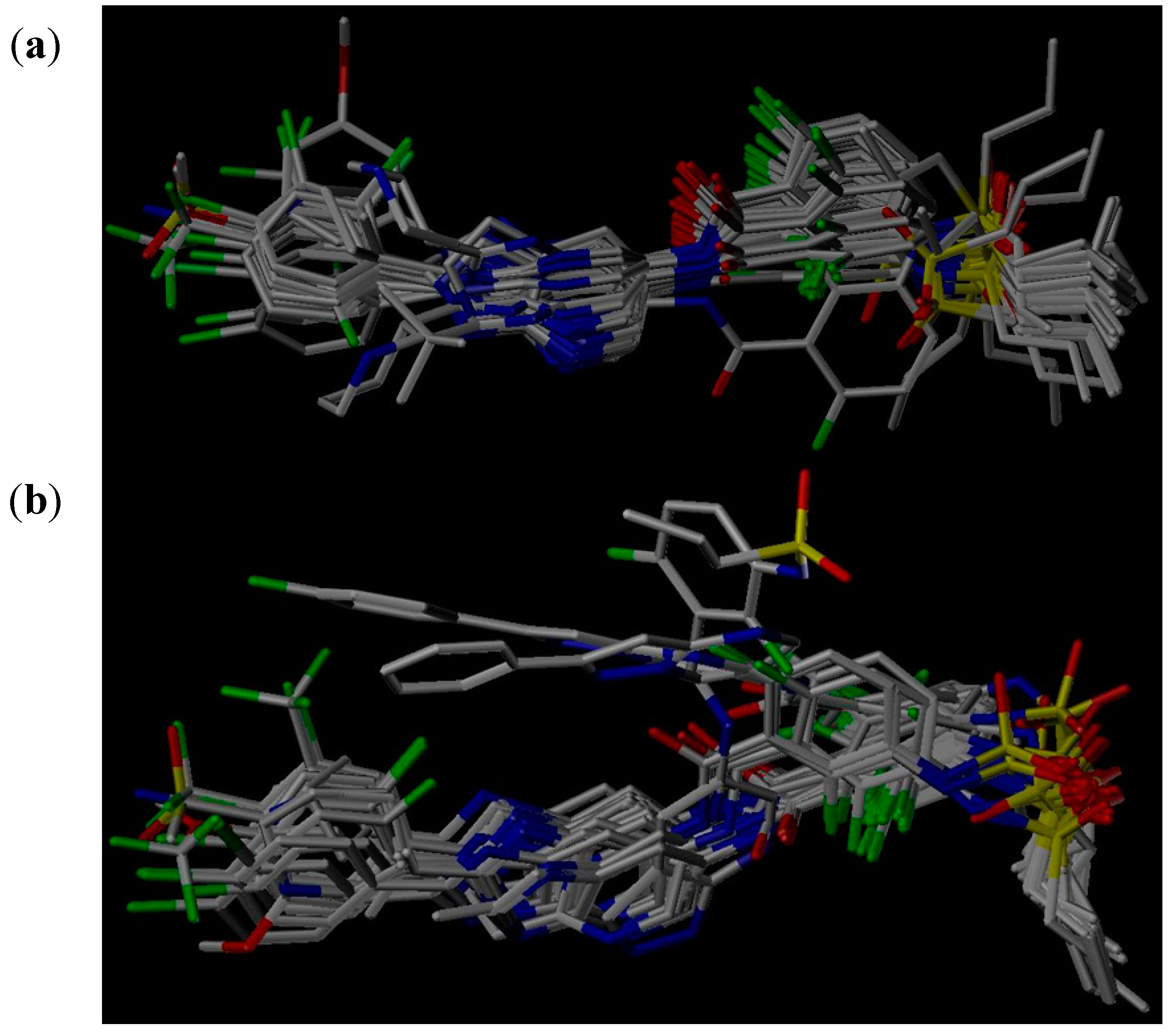
3.5. Molecular Alignment
3.6. CoMFA and CoMSIA Models
3.7. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- El-Azab, A.S.; Al-Omar, M.A.; Abdel-Aziz, A.M.; Abdel-Aziz, N.I.; El-Sayed, A.A.; Aleisa, A.M.; Sayed-Ahmed, M.M.; Abdel-Hamide, S.G. Design, synthesis and biological evaluation of novel quinazoline derivatives as potential antitumor agents: Molecular docking study. Eur. J. Med. Chem. 2010, 45, 4188–4198. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batt, D.; Warmuth, M. B-Raf kinase inhibitors for cancer treatment. Curr. Opin. Investig. Drugs 2007, 8, 452–456. [Google Scholar] [PubMed]
- Hoshino, R.; Chantani, Y.; Yamori, T.; Tsuruo, T.; Oka, H.; Yoshida, O.; Shimada, Y.; Ari-i, S.; Wada, H.; Fujimoto, J.; et al. Constitutive activation of the 41-/43-kDa mitogen-activated protein kinase signaling pathway in human tumors. Oncogene 1999, 18, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Mercer, K.E.; Pritchard, C.A. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim. Biophys. Acta 2003, 1653, 25–40. [Google Scholar] [CrossRef]
- Tuveson, D.A.; Weber, B.L.; Herlyn, M. BRAF as a potential therapeutic target in melanoma and other malignancies. Cancer Cell 2003, 4, 95–98. [Google Scholar] [CrossRef]
- Karasarides, M.; Chiloeches, A.; Hayward, R.; Niculescu-Duvaz, D.; Scanlon, I.; Friedlos, F.; Ogilvie, L.; Hedley, D.; Martin, J.; Marshall, C.J.; et al. B-RAF is a therapeutic target in melanoma. Oncogene 2004, 23, 6292–6298. [Google Scholar] [CrossRef] [PubMed]
- Garnett, M.J.; Marais, R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell 2004, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Madhunapantula, S.V.; Robertson, G.P. Is B-Raf a good therapeutic target for melanoma and other malignancies? Cancer Res. 2008, 68, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Zhang, H.; Zheng, X.; Gao, D. 3D QSAR studies on a series of potent and high selective inhibitors for three kinases of RTK family. J. Mol. Graph. Model. 2007, 26, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Ma, Y.; Wang, R.L.; Xu, W.R.; Wang, S.Q.; Chou, K.C. Find novel dual-agonist drugs for treating type 2 diabetes by means of cheminformatics. Drug Des. Dev. Ther. 2013, 7, 279–287. [Google Scholar]
- Bhatt, H.G.; Patel, P.K. Pharmacophore modeling, virtual screening and 3D-QSAR studies of 5-tetrahydroquinolinylidine aminoguanidine derivatives as sodium hydrogen exchanger inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 3758–3765. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.S.; Huang, R.B.; Wei, Y.T. Multiple field three dimensional quantitative structure-activity relationship (MF-3D-QSAR). J. Comput. Chem. 2008, 29, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Cichero, E.; Fossa, P. Docking-based 3D-QSAR analyses of pyrazole derivatives as HIV-1 non-nucleoside reverse transcriptase inhibitors. J. Mol. Model. 2012, 18, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Z.; Liu, Y.J.; Hu, S.Y.; Zhang, H.B. 3D-QSAR study of Chk1 kinase inhibitors based on docking. J. Mol. Model. 2012, 18, 3669–3694. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yu, R.; Shen, B.Z.; Xu, Y.; Liu, Y.F.; Zheng, H.; Yao, W.B. Docking-based 3D-QSAR modeling of the inhibitors of IMP metallo-β-lactamase. Med. Chem. Res. 2013, 22, 1730–1739. [Google Scholar] [CrossRef]
- Putz, M.V. Residual-QSAR: Implications for genotoxic carcinogenesis. Chem. Cent. J. 2011, 5. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Ionaşcu, C.; Putz, A.M.; Ostafe, V. Alert-QSAR: Implications for electrophilic theory of chemical carcinogenesis. Int. J. Mol. Sci. 2011, 12, 5098–5134. [Google Scholar] [CrossRef] [PubMed]
- Putz, M.V.; Dudaş, N.A. Variational principles for mechanistic quantitative structure-activity relationship (QSAR) studies: Application on uracil derivatives’ anti-HIV action. Struct. Chem. 2013, 24, 1873–1893. [Google Scholar] [CrossRef]
- Putz, M.V.; Dudaş, N.A. Determining chemical reactivity driving biological activity from smiles transformations: The bonding mechanism of anti-HIV pyrimidines. Molecules 2013, 18, 9061–9116. [Google Scholar] [CrossRef] [PubMed]
- Cramer, R.D.; Patterson, D.E.; Bunce, J.D. Comparative molecular field analysis (CoMFA). 1. Effect of shape on binding of steroids to carrier proteins. J. Am. Chem. Soc. 1988, 110, 5959–5967. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130–4146. [Google Scholar] [CrossRef] [PubMed]
- Newhouse, B.J.; Wenglowsky, S.; Grina, J.; Laird, E.R.; Voegtli, W.C.; Ren, L.; Ahrendt, K.; Buckmelter, A.; Gloor, S.L.; Klopfenstein, N.; et al. Imidazo[4,5-b]pyridine inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2013, 23, 5896–5899. [Google Scholar] [CrossRef] [PubMed]
- Dorfman, R.J.; Smith, K.M.; Masek, B.B.; Clark, R.D. A knowledge-based approach to generating diverse but energetically representative ensembles of ligand conformers. J. Comput. Aided Mol. Des. 2008, 22, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010, 29, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Kothandan, G.; Madhavan, T.; Gadhe, C.G.; Cho, S.J. A combined 3D QSAR and pharmacophore-based virtual screening for the identification of potent p38 MAP kinase inhibitors: an in silico approach. Med. Chem. Res. 2013, 22, 1773–1787. [Google Scholar] [CrossRef]
- Xie, H.; Qiu, K.; Xie, X. 3D QSAR studies, pharmacophore modeling and virtual screening on a series of steroidal aromatase inhibitors. Int. J. Mol. Sci. 2014, 15, 20927–20947. [Google Scholar] [CrossRef] [PubMed]
- Thaimattam, R.; Daga, P.R.; Banerjee, R.; Iqbal, J. 3D-QSAR studies on c-Src kinase inhibitors and docking analyses of a potent dual kinase inhibitor of c-Src and c-Abl kinases. Bioorg. Med. Chem. 2005, 13, 4704–4712. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.J.; Barbault, F.; Delamar, M.; Zhang, R.S. Receptor- and ligand-based 3D-QSAR study for a series of non-nucleoside HIV-1 reverse transcriptase inhibitors. Bioorg. Med. Chem. 2009, 17, 2400–2409. [Google Scholar] [CrossRef] [PubMed]
- Richmond, N.J.; Abrams, C.A.; Wolohan, P.R.N.; Abrahamian, E.; Willett, P.; Clark, R.D. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J. Comput. Aided Mol. Des. 2006, 20, 567–587. [Google Scholar] [CrossRef] [PubMed]
- Shepphird, J.K.; Clark, R.D. A marriage made in torsional space: Using GALAHAD models to drive pharmacophore multiplet searches. J. Comput. Aided Mol. Des. 2006, 20, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.H.; Salum, L.B.; Pasqualoto, K.F.M.; Ferreira, E.I.; Andricopulo, A.D. Three-dimensional quantitative structure-activity relationships for a large series of potent antitubercular agents. Lett. Drug Des. Discov. 2008, 5, 377–387. [Google Scholar] [CrossRef]
- Wang, J.N.; Wang, F.F.; Xiao, Z.T.; Sheng, G.W.; Li, Y.; Wang, Y.H. Molecular simulation of a series of benzothiazole PI3K α inhibitors: Probing the relationship between structural features, anti-tumor potency and selectivity. J. Mol. Model. 2012, 18, 2943–2958. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Chen, L.; Zhang, J.; Xie, X.; Qiu, K.; Fu, J. A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors. Int. J. Mol. Sci. 2015, 16, 12307-12323. https://doi.org/10.3390/ijms160612307
Xie H, Chen L, Zhang J, Xie X, Qiu K, Fu J. A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors. International Journal of Molecular Sciences. 2015; 16(6):12307-12323. https://doi.org/10.3390/ijms160612307
Chicago/Turabian StyleXie, Huiding, Lijun Chen, Jianqiang Zhang, Xiaoguang Xie, Kaixiong Qiu, and Jijun Fu. 2015. "A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors" International Journal of Molecular Sciences 16, no. 6: 12307-12323. https://doi.org/10.3390/ijms160612307
APA StyleXie, H., Chen, L., Zhang, J., Xie, X., Qiu, K., & Fu, J. (2015). A Combined Pharmacophore Modeling, 3D QSAR and Virtual Screening Studies on Imidazopyridines as B-Raf Inhibitors. International Journal of Molecular Sciences, 16(6), 12307-12323. https://doi.org/10.3390/ijms160612307