
Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells

Abstract
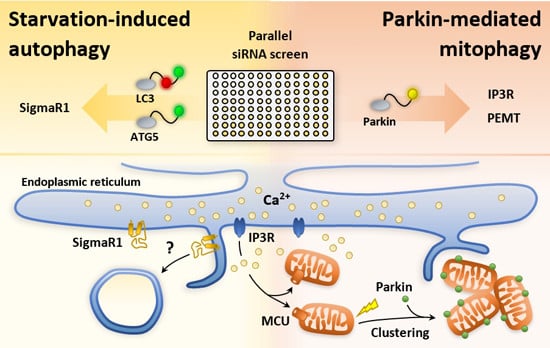
:
1. Introduction
2. Results
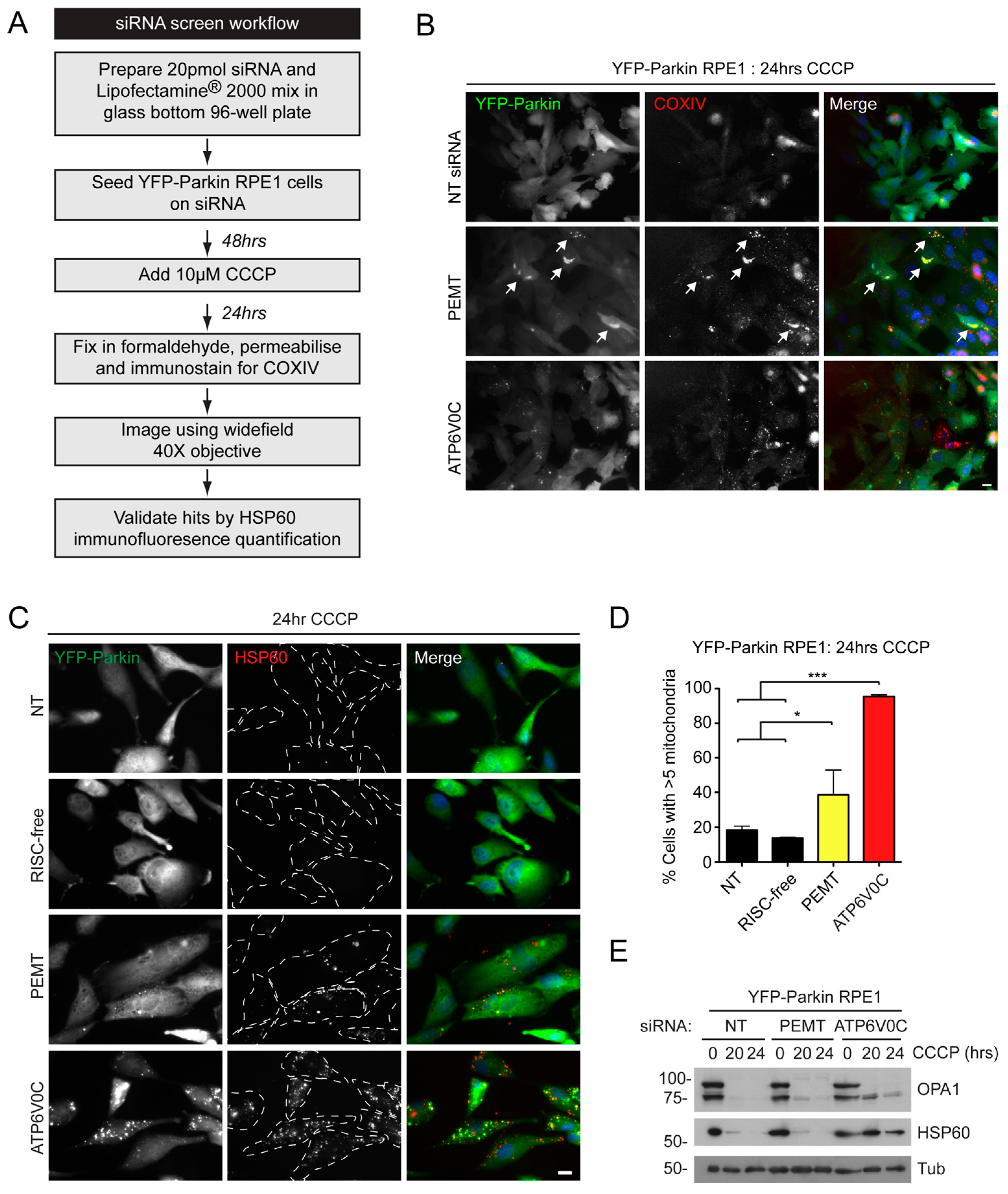
2.1. An siRNA Screen for Mitochondrial/Endoplasmic Reticulum (ER) Based Mediators of Starvation-Induced Autophagy Identifies SIGMAR1

{kind=link}
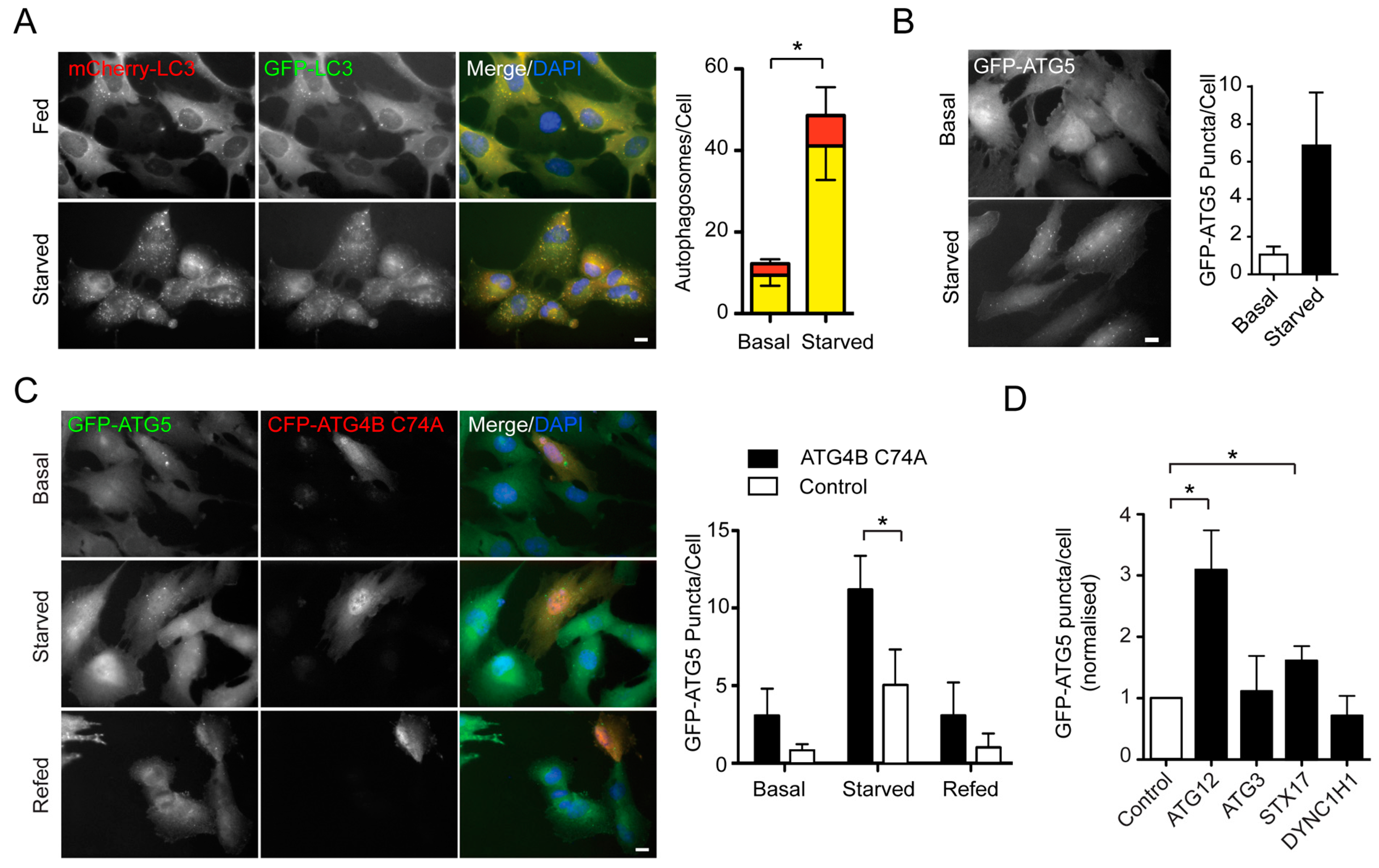{kind=link}
{kind=link}
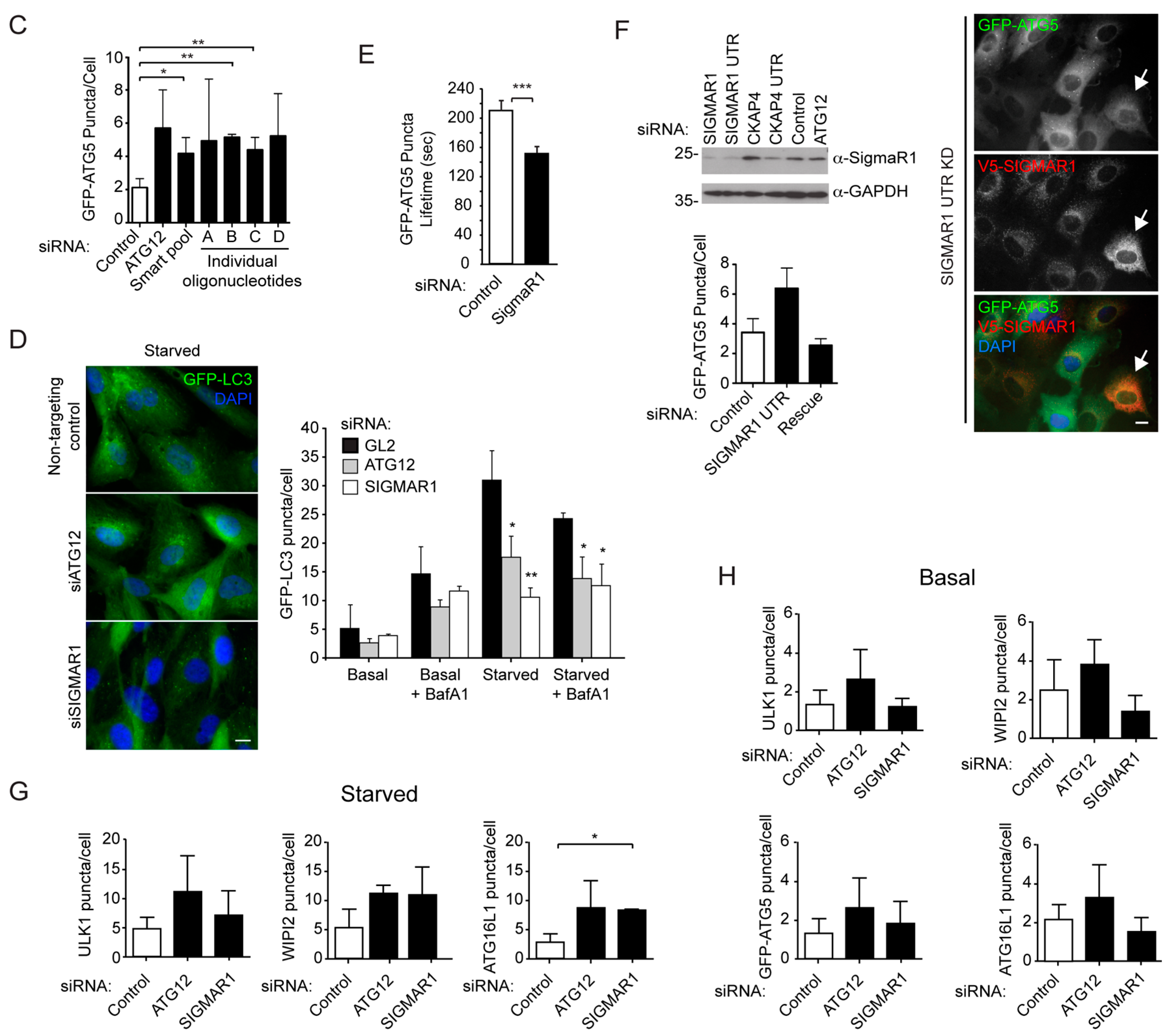{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Gene ID | siRNA Cat. No | Autophagy Screen | Mitophagy Inhibited? | ||
|---|---|---|---|---|---|---|
| ATG5 | LC3 | Defect? | ||||
| DNM1L | 10059 | M-012092-01 | 0.6 | 0.7 | No | No |
| HSPA9 | 3313 | M-004750-03 | 1.4 | 1.05 | No | No |
| MFF | 56947 | M-018261-01 | 1.7 | 0.6 | No | No |
| MARCH5 | 54708 | M-007001-0 | 0.65 | 0.6 | No | No |
| FIS1 | 51024 | M-020907-02 | 0.85 | 0.3 | No | No |
| MFN1 | 55669 | M-010670-01 | 1.0 | 0.4 | No | No |
| MFN2 | 9927 | M-012961-00 | 1.0 | 0.15 | No | No |
| RHOT1 | 55288 | D-010365-01 | 0.85 | 0.85 | No | ND |
| RHOT2 | 89941 | M-008340-01 | 0.65 | 0.4 | No | ND |
| BCAP31 | 10134 | M-018679-00 | 1.4 | 0.1 | No | No |
| PACS2 | 23241 | M-022015-01 | 0.65 | 0.3 | No | No |
| TRAK1 | 22906 | M-020331-02 | 0.8 | 0.5 | No | ND |
| TRAK2 | 66008 | M-014141-00 | 1.2 | 0.4 | No | ND |
| VAPB | 9217 | M-017795-00 | 1.85 | 0.35 | Yes | No |
| STX18 | 53407 | M-020624-00 | 1.05 | 0.05 | No | No |
| VDAC1 | 7416 | M-019764-00 | 0.95 | 0.25 | No | No |
| IP3R1 | 3708 | M-006207-01 | 0.3 | 0.85 | No | No |
| IP3R2 | 3709 | M-006208-01 | 0.55 | 0.55 | No | No |
| IP3R3 | 38710 | M-006209-02 | 0.55 | 0.8 | No | No |
| ATF6 | 22926 | M-009917-01 | 1.05 | 0.55 | No | No |
| EIF3AK3 | 9451 | M-004883-03 | 0.55 | 0.7 | No | No |
| ERN1 | 2081 | M-004951-02 | 0.5 | 1.1 | No | No |
| ANK2 | 287 | M-008417-02 | 0.65 | 1.4 | No | No |
| ATP2A1 | 487 | M-006113-00 | 1.4 | 0.3 | No | No |
| ATP2A2 | 488 | M-004082-01 | 1.9 | 0.4 | Yes | No |
| ATP2A3 | 489 | M-006114-01 | 0.85 | 0.75 | No | No |
| RMDN3 | 55177 | D-020973-01 | 0.7 | 0.35 | No | No |
| TMBIM6 | 7009 | M-004118-01 | 0.9 | 2.1 | No | No |
| ESYT1 | 23344 | M-010652-00 | 1.3 | 1.35 | No | No |
| ESYT2 | 57488 | M-025231-00 | 0.8 | 1.35 | No | No |
| ESYT3 | 83850 | M-023602-01 | 0.55 | 0.45 | No | No |
| SIGMAR1 | 10280 | M-017475-02 | 2.3 | 0.2 | Yes | No |
| PEMT | 10400 | D-010392-01 | 0.85 | 0.8 | No | Yes |
| PISD | 23761 | M-009548-00 | 1.6 | 0.85 | No | No |
| PTDSS1 | 9791 | M-008568-00 | 0.65 | 0.4 | No | No |
| PTDSS2 | 81490 | M-008959-00 | 0.95 | 0.7 | No | No |
| TEX2 | 55852 | M-017117-00 | 0.9 | 1.15 | No | No |
| ATL2 | 64225 | M-014047-00 | 0.65 | 0.65 | No | No |
| ATL3 | 25923 | M-010656-00 | 0.15 | 1.35 | No | No |
| CKAP4 | 10970 | M-012755-01 | 1.5 | 0.25 | Yes | No |
| KTN1 | 3895 | M-010605-01 | 0.55 | 0.95 | No | No |
| REEP1 | 65055 | M-014235-01 | 1.45 | 0.75 | No | No |
| REEP2 | 51308 | M-021201-00 | 0.85 | 0.2 | No | No |
| REEP3 | 221035 | M-032301-0 | 0.85 | 0.9 | No | No |
| REEP5 | 92840 | M-019467-0 | 0.4 | 0.3 | No | No |
| REEP6 | 7905 | M-015555-00 | 0.4 | 0.3 | No | No |
| RTN1 | 6252 | M-014138-00 | 1.4 | 1.1 | No | No |
| RTN2 | 6253 | M-012717-00 | 0.95 | 0.35 | No | No |
| RTN3 | 10313 | M-020088-00 | 0.9 | 1.1 | No | No |
| RTN4 | 57142 | M-010721-00 | 1.1 | 0.45 | No | No |
| PSEN2 | 5664 | M-006018-02 | 1.1 | 0.55 | No | No |
| LRPPRC | 10128 | M-018773-00 | 0.45 | 0.75 | No | No |
| MAPRE1 | 22919 | M-006824-00 | 1.0 | 1.3 | No | No |
| PDZD8 | 118987 | M-018369-01 | 0.85 | 0.4 | No | No |
| SPAST | 6683 | M-014070-02 | 1.55 | 0.45 | Yes | No |
| TCHP | 84260 | M-014843-01 | 1.1 | 1.1 | No | No |
| APC | 324 | M-003869-01 | 0.45 | 0.96 | No | No |
| BAX | 581 | M-003308-03 | 0.25 | 0.2 | No | No |
| BAK1 | 578 | M-003305-02 | 1.0 | 0.25 | No | No |
| BCL2 | 596 | M-003307-06 | 0.3 | 0.5 | No | No |
| MAP1S | 55201 | M-016881-01 | 0.55 | 0.65 | No | No |
| ATG12 | 9140 | N/A | 2.55 | 0.15 | Yes | ND |
| STX17 | 55014 | 10620318/9 | 1.55 | 0.7 | No | ND |
| MEAN | - | N/A | 0.93 | 0.67 | N/A | N/A |



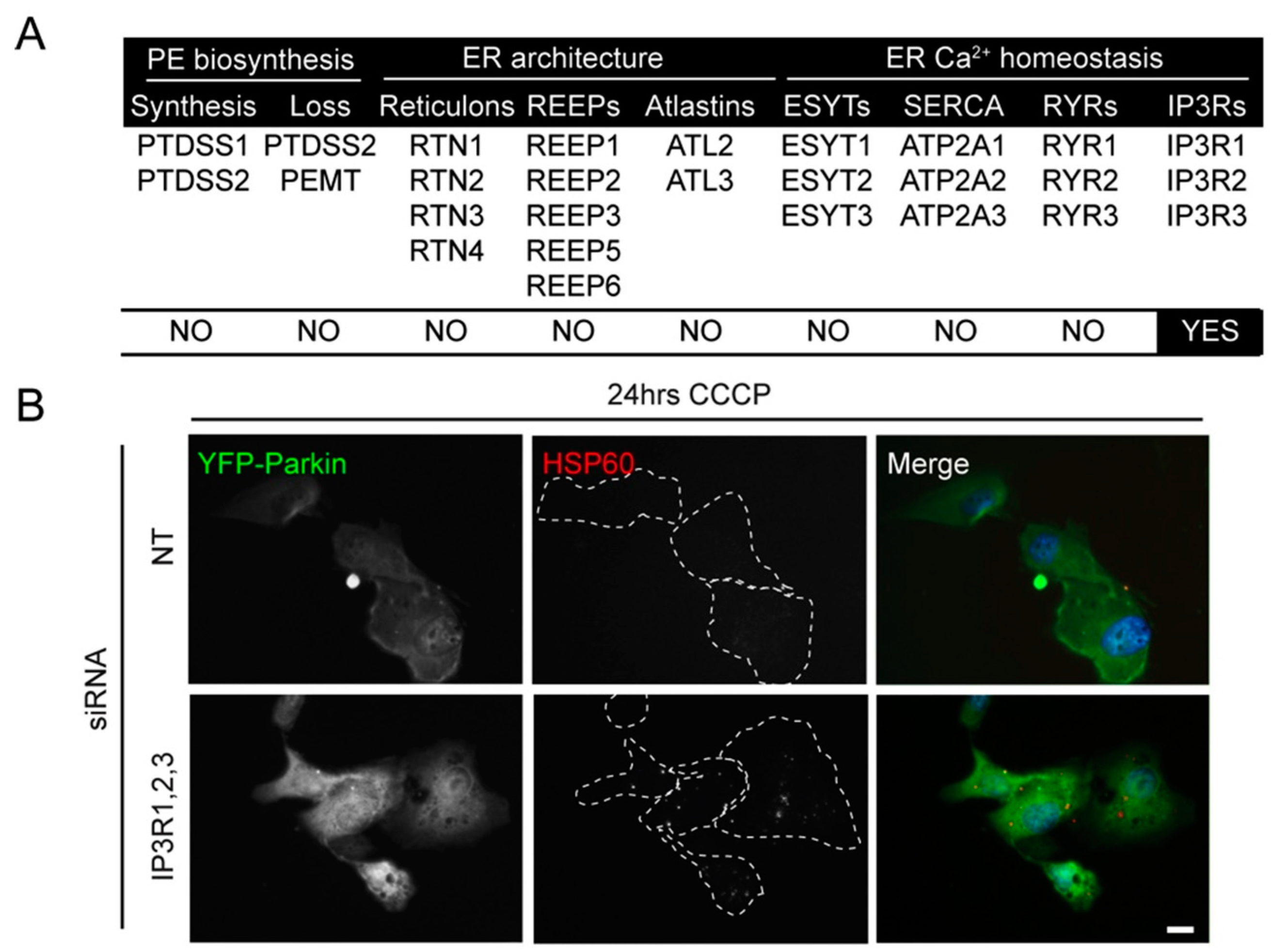
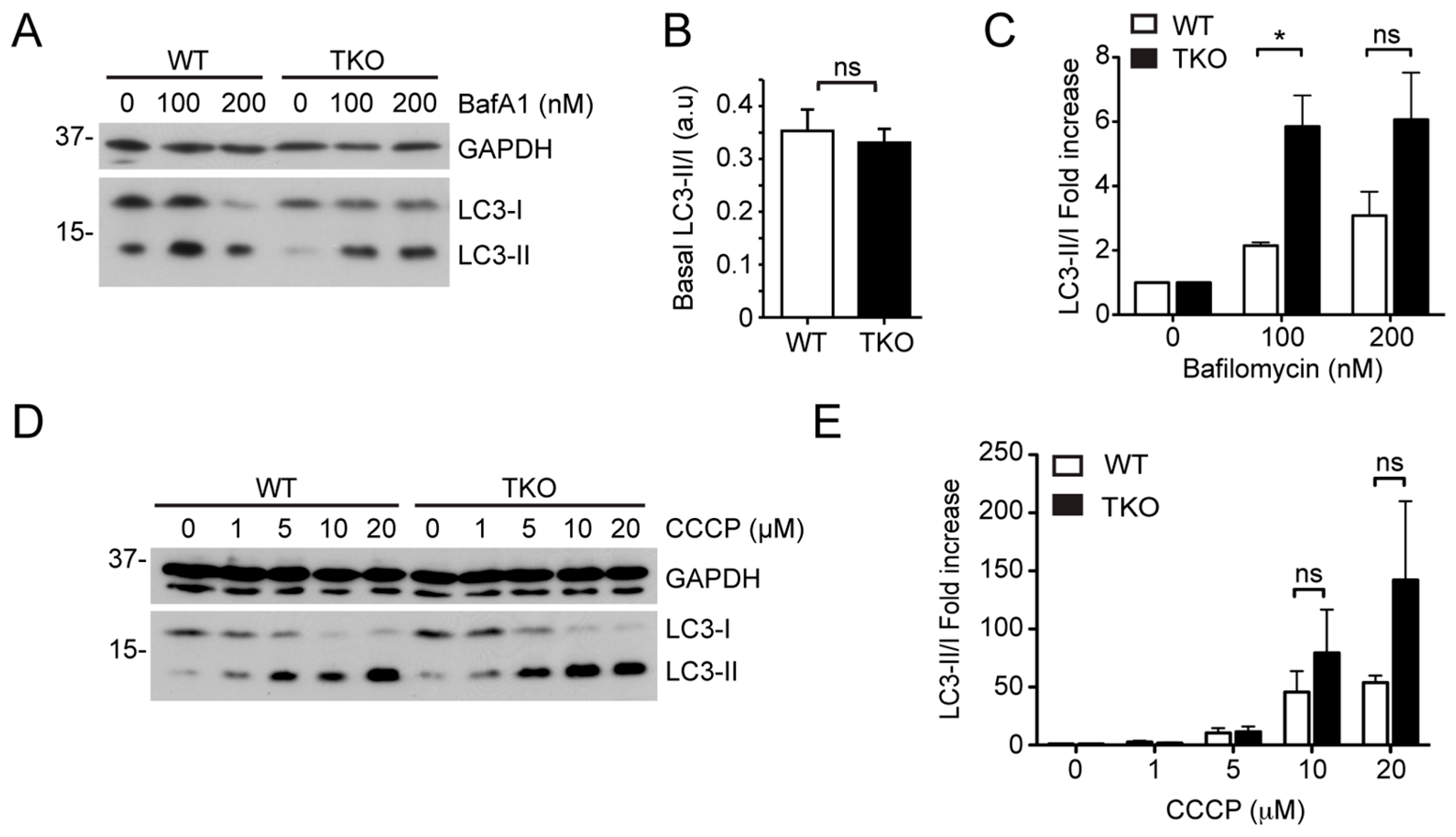
2.2. siRNA Screens Identify Lipid Biogenesis and Ca2+ Homeostasis Regulators as Mediators of Parkin-Mediated Mitophagy



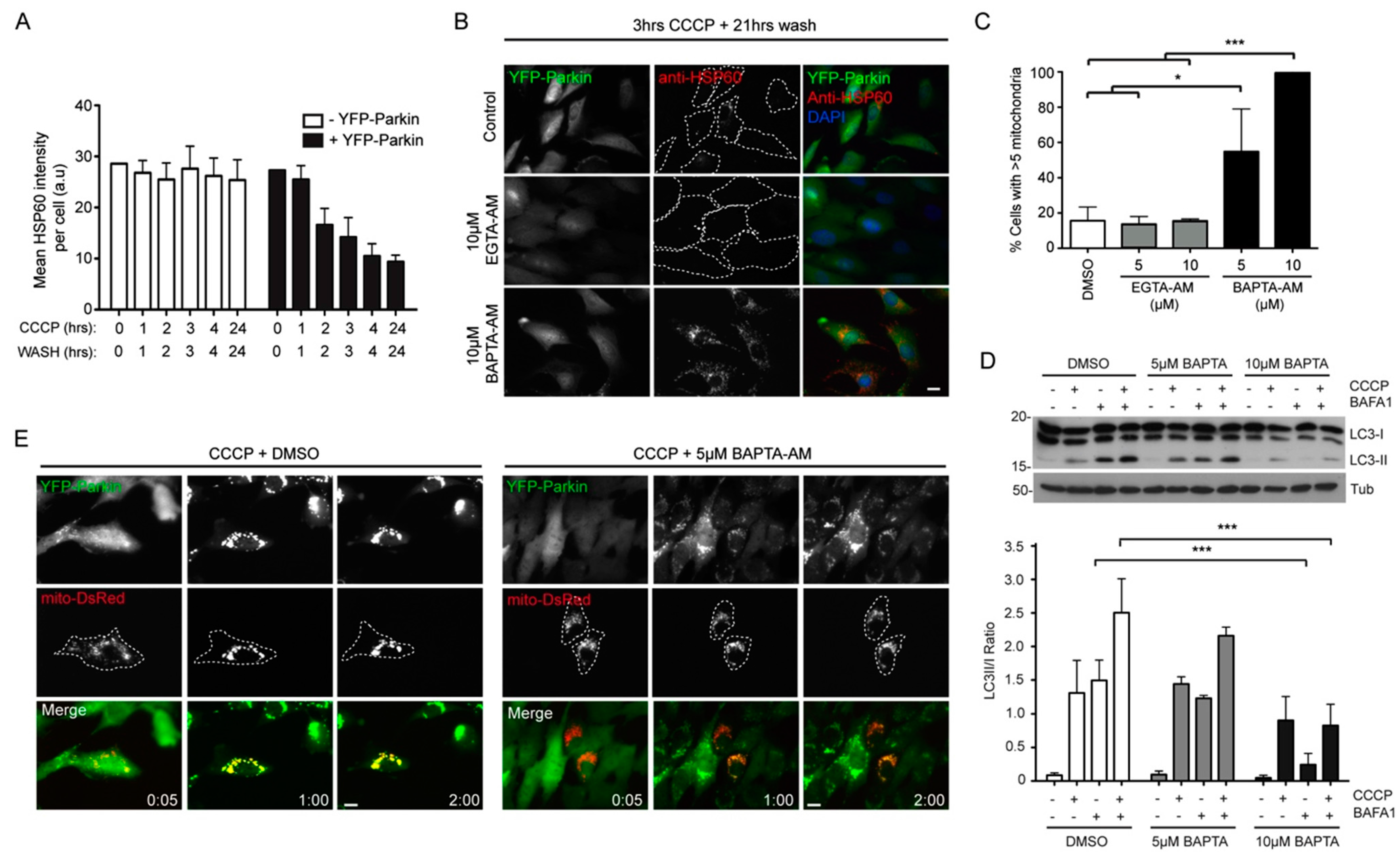
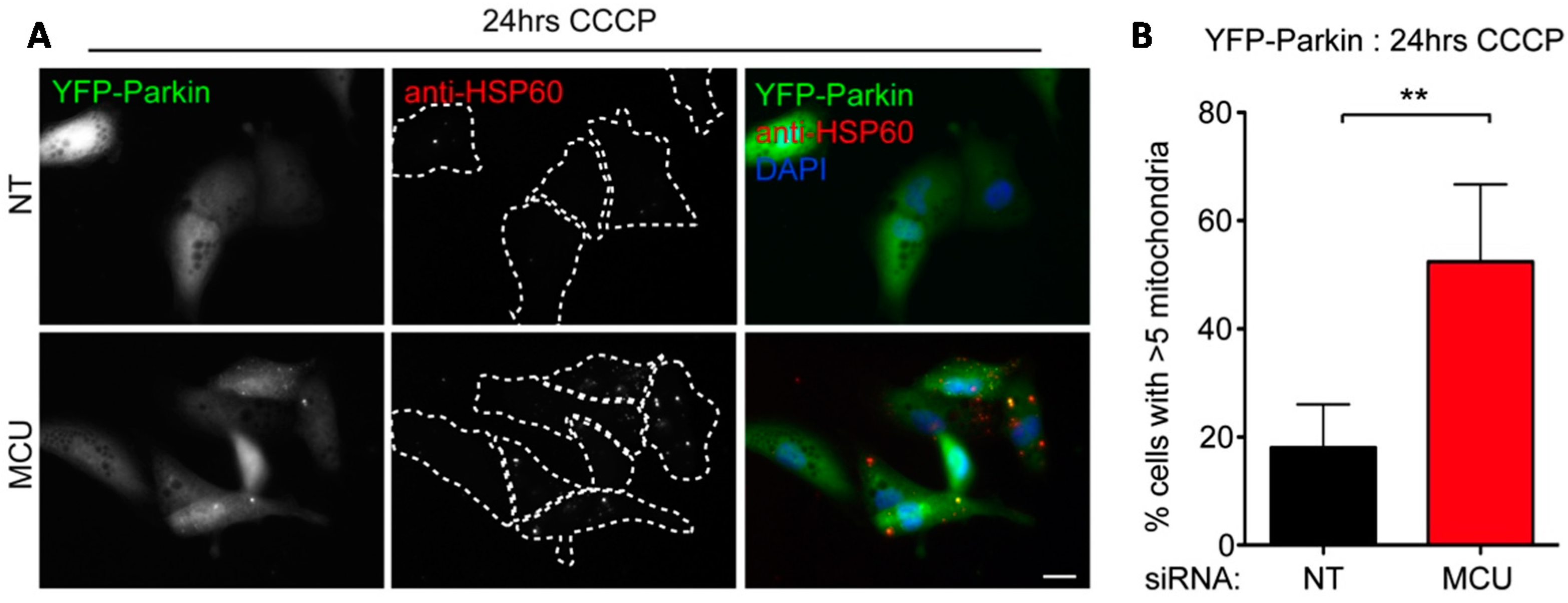
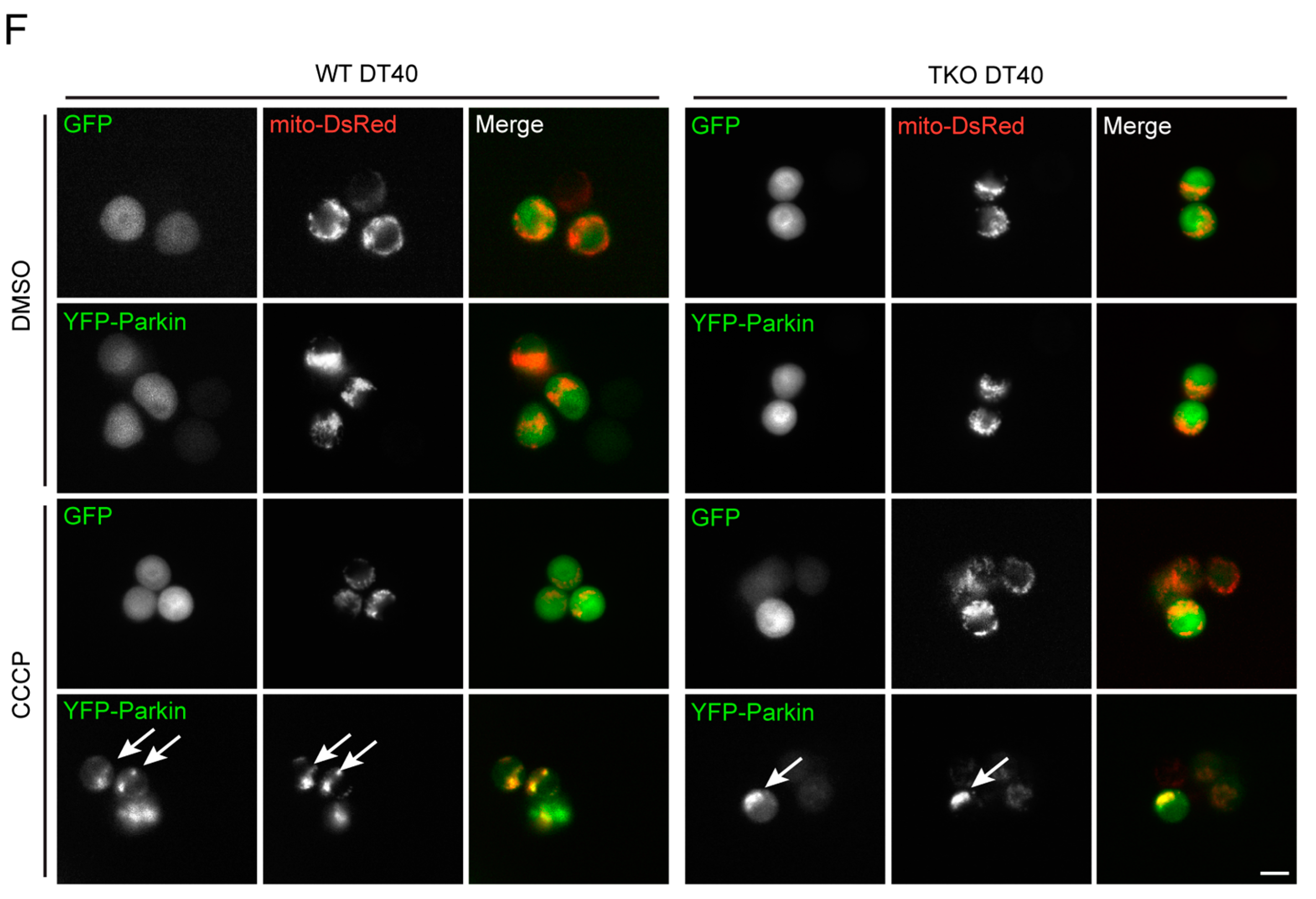
2.3. Local Elevated Cytosolic Ca2+ Is Needed for Efficient Parkin-Mediated Mitophagy




3. Discussion
4. Experimental Section
4.1. Chemicals and Antibodies
4.2. Cell Culture and Cell-Lines
4.3. Transfection and Viral Transduction
4.4. siRNA Library and Transfection
4.5. Microscopy
Acknowledgments
Author Contributions
Conflicts of Interest
References
- MacVicar, T.D.B. Mitophagy. Essays Biochem. 2013, 55, 93–104. [Google Scholar]
- MacVicar, T.D.B.; Lane, J.D. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J. Cell Sci. 2014, 127, 2313–2325. [Google Scholar] [PubMed]
- Tooze, S.A.; Yoshimori, T. The origin of the autophagosomal membrane. Nat. Cell Biol. 2010, 12, 831–835. [Google Scholar] [PubMed]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [PubMed]
- Hayashi-Nishino, M.; Fujita, N.; Noda, T.; Yamaguchi, A.; Yoshimori, T.; Yamamoto, A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009, 11, 1433–1437. [Google Scholar] [CrossRef] [PubMed]
- Yla-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy 2009, 5, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Koyama-Honda, I.; Mizushima, N. Structures containing ATG9A and the ULK1 complex independently target depolarized mitochondria at initial stages of parkin-mediated mitophagy. J. Cell Sci. 2012, 125, 1488–1499. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Van Vliet, A.R.; Verfaillie, T.; Agostinis, P. New functions of mitochondria associated membranes in cellular signaling. Biochim. Biophys. Acta 2014, 1843, 2253–2262. [Google Scholar] [CrossRef] [PubMed]
- Celsi, F.; Pizzo, P.; Brini, M.; Leo, S.; Fotino, C.; Pinton, P.; Rizzuto, R. Mitochondria, calcium and cell death: A deadly triad in neurodegeneration. Biochim. Biophys. Acta 2009, 1787, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial atp synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef] [PubMed]
- Tinel, H.; Cancela, J.M.; Mogami, H.; Gerasimenko, J.V.; Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J. 1999, 18, 4999–5008. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Macaskill, A.F.; Rinholm, J.E.; Twelvetrees, A.E.; Arancibia-Carcamo, I.L.; Muir, J.; Fransson, A.; Aspenstrom, P.; Attwell, D.; Kittler, J.T. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 2009, 61, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.T.; Niescier, R.F.; Min, K.T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. USA 2011, 108, 15456–15461. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Schwarz, T.L. The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 2009, 136, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Saotome, M.; Safiulina, D.; Szabadkai, G.; Das, S.; Fransson, A.; Aspenstrom, P.; Rizzuto, R.; Hajnoczky, G. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the MIRO GTPASE. Proc. Natl. Acad. Sci. USA 2008, 105, 20728–20733. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; de Smedt, H.; Parys, J.B.; Bultynck, G. IP3 receptors, mitochondria, and ca signaling: Implications for aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef] [PubMed]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Monkawa, T.; Miyawaki, A.; Sugiyama, T.; Yoneshima, H.; Yamamoto-Hino, M.; Furuichi, T.; Saruta, T.; Hasegawa, M.; Mikoshiba, K. Heterotetrameric complex formation of inositol 1,4,5-trisphosphate receptor subunits. J. Biol. Chem. 1995, 270, 14700–14704. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I.; Watras, J.; Ehrlich, B.E. Bell-shaped calcium-response curves of ins(1,4,5)p3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 1991, 351, 751–754. [Google Scholar] [CrossRef] [PubMed]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Criollo, A.; Maiuri, M.C.; Tasdemir, E.; Vitale, I.; Fiebig, A.A.; Andrews, D.; Molgo, J.; Diaz, J.; Lavandero, S.; Harper, F.; et al. Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Differ. 2007, 14, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive INSP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Joseph, S.K. Role of inositol trisphosphate receptors in autophagy in DT40 cells. J. Biol. Chem. 2010, 285, 16912–16920. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. Pink1 is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed]
- Backer, J.M. The regulation and function of class III PI3KS: Novel roles for VPS34. Biochem. J. 2008, 410, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Hayashi-Nishino, M.; Fukumoto, H.; Omori, H.; Yamamoto, A.; Noda, T.; Yoshimori, T. An ATG4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell 2008, 19, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the ATG5-ATG12/ATG16 complex during autophagosome formation. EMBO J. 2012, 31, 4304–4317. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Kuma, A.; Kobayashi, Y.; Yamamoto, A.; Matsubae, M.; Takao, T.; Natsume, T.; Ohsumi, Y.; Yoshimori, T. Mouse APG16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the APG12-APG5 conjugate. J. Cell Sci. 2003, 116, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Rojas-Rivera, D.; Lisbona, F.; Caballero, B.; Nassif, M.; Court, F.A.; Schuck, S.; Ibar, C.; Walter, P.; Sierralta, J.; et al. Bax inhibitor-1 regulates autophagy by controlling the IRE1α branch of the unfolded protein response. EMBO J. 2011, 30, 4465–4478. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Alemu, E.A.; Lamark, T.; Torgersen, K.M.; Birgisdottir, A.B.; Larsen, K.B.; Jain, A.; Olsvik, H.; Overvatn, A.; Kirkin, V.; Johansen, T. ATG8 family proteins act as scaffolds for assembly of the ulk complex: Sequence requirements for LC3-interacting region (LIR) motifs. J. Biol. Chem. 2012, 287, 39275–39290. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Girardi, J.P.; Bakovic, M. Forms, crosstalks, and the role of phospholipid biosynthesis in autophagy. Int. J. Cell Biol. 2012, 2012, 931956. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, L.; Li, S.; Sheng, L.; Cao, Z.; Chen, L. Expressions of protease-activated receptors in human gingival fibroblasts and its functions in periodontitis. Chin. J. Stomatol. 2014, 49, 21–26. [Google Scholar]
- Tsien, R.Y. New calcium indicators and buffers with high selectivity against magnesium and protons: Design, synthesis, and properties of prototype structures. Biochemistry 1980, 19, 2396–2404. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, E.; Bucurenciu, I.; Goswami, S.P.; Jonas, P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat. Rev. Neurosci. 2012, 13, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef] [PubMed]
- 4Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. Micu1 is an essential gatekeeper for mcu-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Tan, Y.W.; Hagenston, A.M.; Martel, M.A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Kindt, D.; Luyten, T.; Welkenhuyzen, K.; Missiaen, L.; de Smedt, H.; Bultynck, G.; Parys, J.B. MTOR-controlled autophagy requires intracellular Ca2+ signaling. PLoS ONE 2013, 8, e61020. [Google Scholar] [CrossRef] [PubMed]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; de Smedt, H.; Parys, J.B.; et al. Ins(1,4,5)p3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, H.; Kurosaki, M.; Takata, M.; Kurosaki, T. Genetic evidence for involvement of type 1, type 2 and type 3 inositol 1,4,5-trisphosphate receptors in signal transduction through the B-cell antigen receptor. EMBO J. 1997, 16, 3078–3088. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed]
- Piquereau, J.; Caffin, F.; Novotova, M.; Lemaire, C.; Veksler, V.; Garnier, A.; Ventura-Clapier, R.; Joubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [PubMed]
- East, D.A.; Campanella, M. Ca2+ in quality control: An unresolved riddle critical to autophagy and mitophagy. Autophagy 2013, 9, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. Enhanced parkin levels favor ER-mitochondria crosstalk and guarantee Ca2+ transfer to sustain cell bioenergetics. Biochim. Biophys. Acta 2013, 1832, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Bond, J.D.; Ingram, V.M. Compromised mitochondrial function leads to increased cytosolic calcium and to activation of map kinases. Proc. Natl. Acad. Sci. USA 1997, 94, 9705–9710. [Google Scholar] [CrossRef] [PubMed]
- Vaur, S.; Sartor, P.; Dufy-Barbe, L. Calcium store depletion induced by mitochondrial uncoupling in prostatic cells. Gen. Physiol. Biophys. 2000, 19, 265–278. [Google Scholar] [PubMed]
- Tanaka, A.; Cleland, M.M.; Xu, S.; Narendra, D.P.; Suen, D.F.; Karbowski, M.; Youle, R.J. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by parkin. J. Cell Biol. 2010, 191, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Szabadkai, G.; Bianchi, K.; Varnai, P.; de Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Axe, E.L.; Walker, S.A.; Manifava, M.; Chandra, P.; Roderick, H.L.; Habermann, A.; Griffiths, G.; Ktistakis, N.T. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008, 182, 685–701. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MacVicar, T.D.B.; Mannack, L.V.J.C.; Lees, R.M.; Lane, J.D. Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells. Int. J. Mol. Sci. 2015, 16, 13356-13380. https://doi.org/10.3390/ijms160613356
MacVicar TDB, Mannack LVJC, Lees RM, Lane JD. Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells. International Journal of Molecular Sciences. 2015; 16(6):13356-13380. https://doi.org/10.3390/ijms160613356
Chicago/Turabian StyleMacVicar, Thomas D. B., Lilith V. J. C. Mannack, Robert M. Lees, and Jon D. Lane. 2015. "Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells" International Journal of Molecular Sciences 16, no. 6: 13356-13380. https://doi.org/10.3390/ijms160613356
APA StyleMacVicar, T. D. B., Mannack, L. V. J. C., Lees, R. M., & Lane, J. D. (2015). Targeted siRNA Screens Identify ER-to-Mitochondrial Calcium Exchange in Autophagy and Mitophagy Responses in RPE1 Cells. International Journal of Molecular Sciences, 16(6), 13356-13380. https://doi.org/10.3390/ijms160613356