
Inferring Invasion History of Red Swamp Crayfish (Procambarus clarkii) in China from Mitochondrial Control Region and Nuclear Intron Sequences

Abstract
:
1. Introduction
2. Results
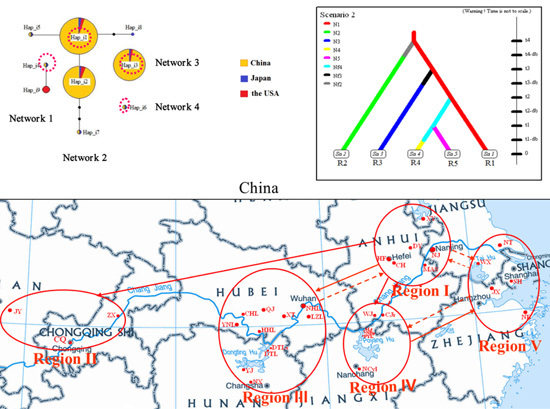
2.1. Genetic Diversity and Haplotypes


{kind=link}
{kind=link}
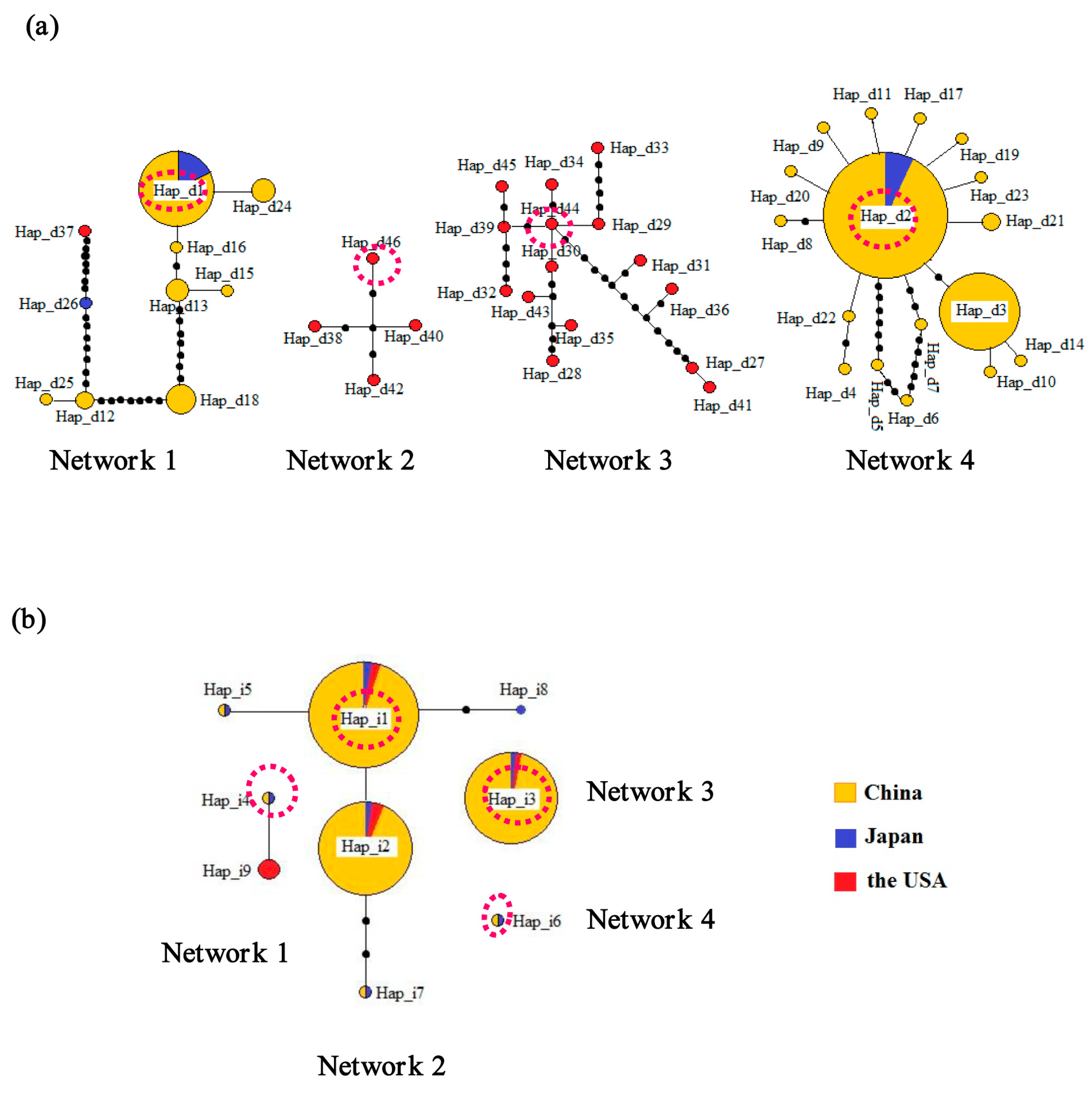{kind=link}
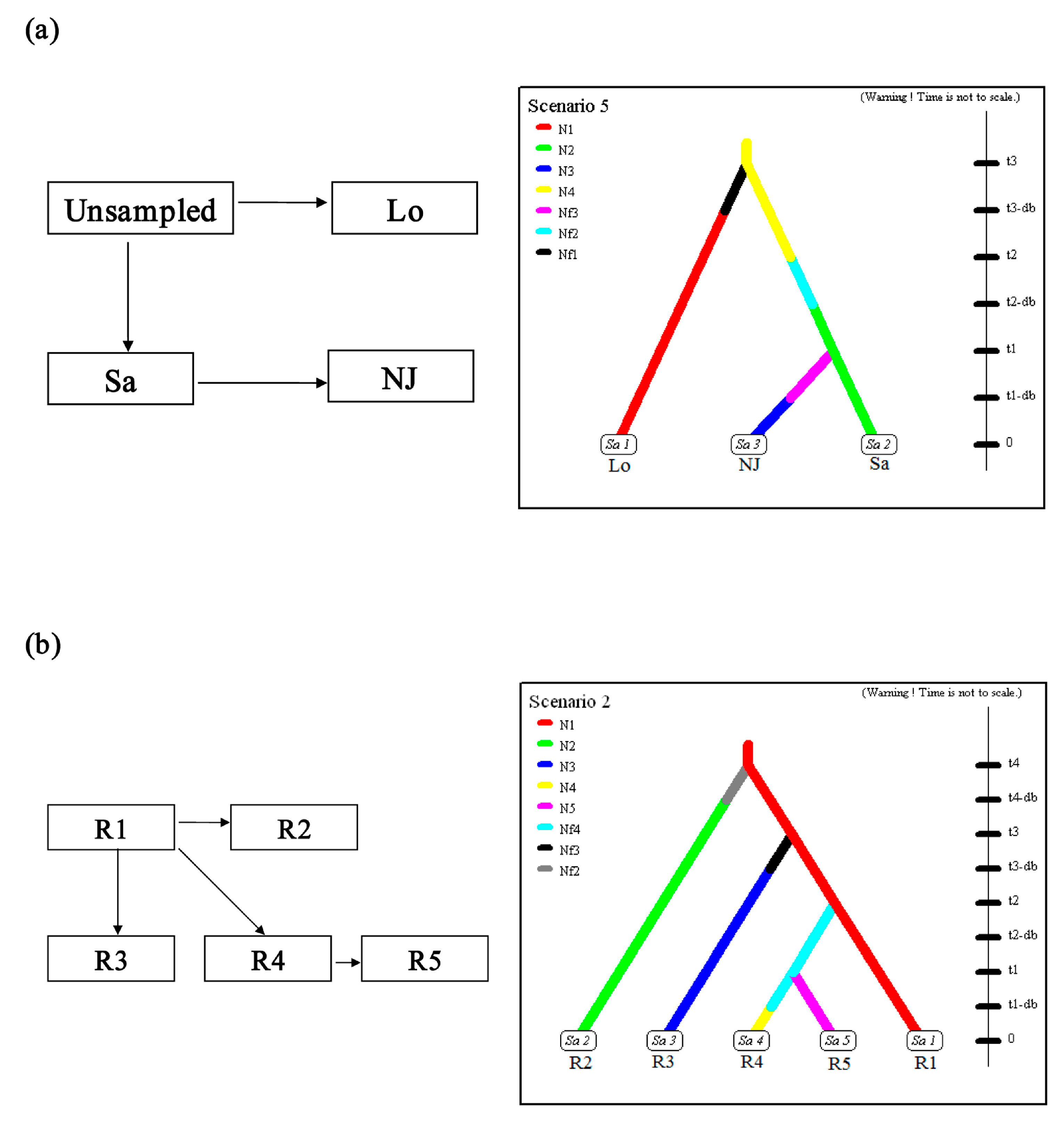{kind=link}
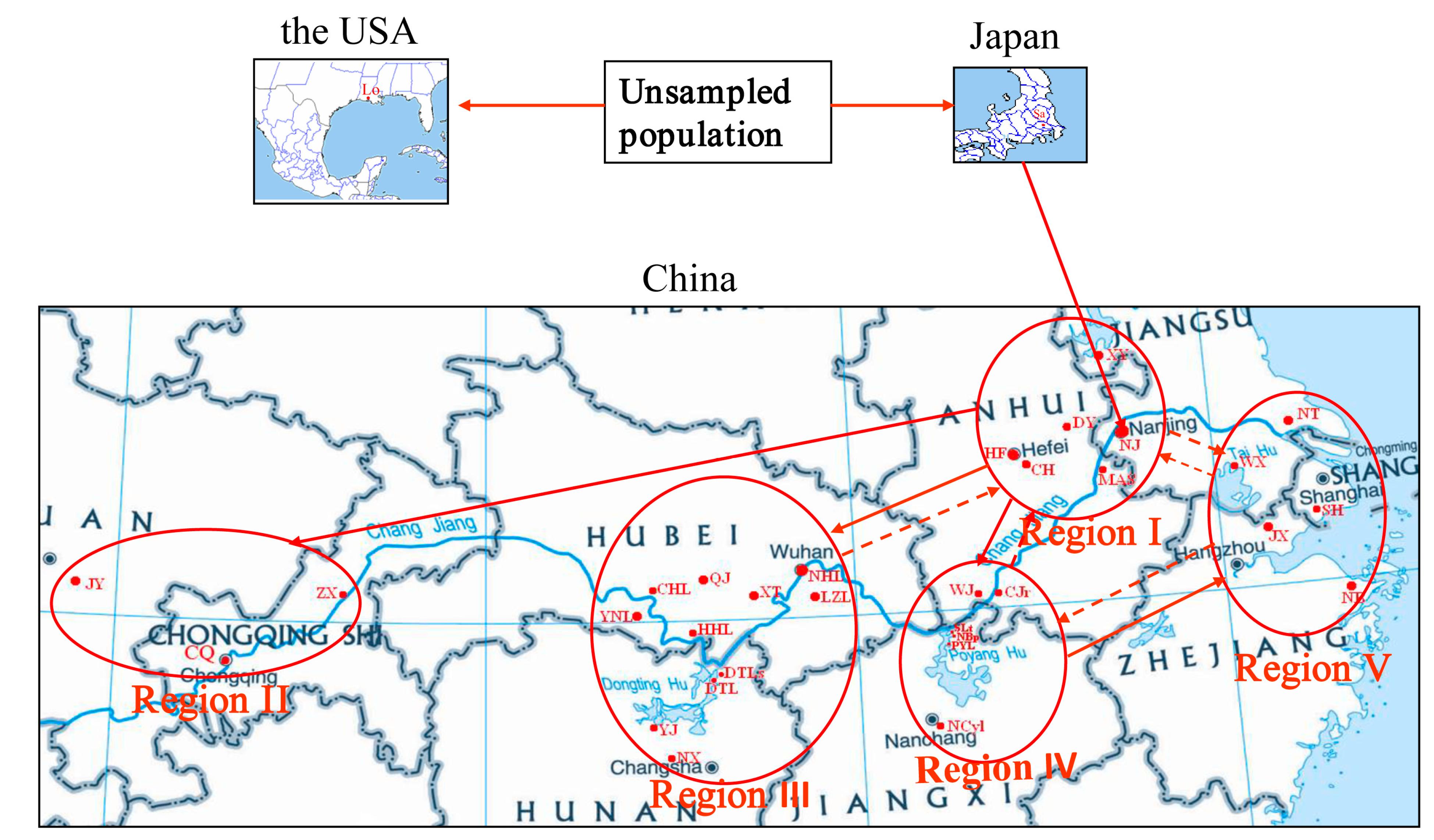{kind=link}
| Code | Location (Country) | Longitude | Latitude | Control Region Sequencing | ProPOx Intron Sequencing | ||||
|---|---|---|---|---|---|---|---|---|---|
| Control Region (N/H) | Hd | Pi | ProPOx Intron (N/H) | Hd | Pi | ||||
| SH | Shanghai (China) | 121.23 °E | 31.03 °N | (7/2) | 0.476 | 0.0121 | (3/3) | 1.000 | 0.0108 |
| NB | Ningbo (China) | 121.55 °E | 29.88 °N | (8/1) | 0.000 | 0.0000 | (4/1) | 0.000 | 0.0000 |
| JX | Jiaxing (China) | 120.77 °E | 30.75 °N | (7/2) | 0.286 | 0.0072 | (2/1) | 0.000 | 0.0000 |
| XYc | Xuyi-culture (China) | 118.50 °E | 33.00 °N | (7/4) | 0.714 | 0.0055 | (6/2) | 0.600 | 0.0097 |
| XYw | Xuyi-wild (China) | 118.42 °E | 33.03 °N | (7/2) | 0.286 | 0.0003 | (4/2) | 0.500 | 0.0020 |
| WXb | Binhu, Wuxi (China) | 120.28 °E | 31.52 °N | (9/2) | 0.500 | 0.0013 | (6/3) | 0.733 | 0.0062 |
| NT | Nantong (China) | 120.87 °E | 32.02 °N | (7/2) | 0.286 | 0.0073 | (6/2) | 0.533 | 0.0065 |
| XG | Xiaguan district (Nanjing, China) | 118.75 °E | 32.08 °N | (8/1) | 0.000 | 0.0000 | (7/2) | 0.476 | 0.0019 |
| XBv | Xiaba village (Nanjing, China) | 118.87 °E | 32.20 °N | (7/1) | 0.000 | 0.0000 | (6/2) | 0.333 | 0.0014 |
| BGt | Baguazhou township (Nanjing, China) | 118.82 °E | 32.17 °N | (7/2) | 0.286 | 0.0003 | (7/2) | 0.476 | 0.0019 |
| WX | Wuxi (China) | 120.30 °E | 31.57 °N | (6/2) | 0.533 | 0.0135 | (7/3) | 0.714 | 0.0081 |
| WJ | Wangjiang (China) | 116.70 °E | 30.12 °N | (7/1) | 0.000 | 0.0000 | (8/2) | 0.536 | 0.0022 |
| MAS | Maanshan (China) | 118.50 °E | 31.55 °N | (8/4) | 0.643 | 0.0114 | (6/2) | 0.333 | 0.0041 |
| CJr | Guangfengwei section of Changjiang river (China) | 116.87 °E | 30.12 °N | (7/2) | 0.286 | 0.0003 | (7/2) | 0.476 | 0.0019 |
| CH | Chaohu (China) | 117.87 °E | 31.62 °N | (7/2) | 0.476 | 0.0121 | (7/2) | 0.476 | 0.0019 |
| HF | Hefei (China) | 117.23 °E | 31.82 °N | (8/2) | 0.250 | 0.0063 | (6/2) | 0.333 | 0.0014 |
| DY | Dingyuan (China) | 117.83 °E | 32.28 °N | (6/2) | 0.600 | 0.0152 | (3/1) | 0.000 | 0.0000 |
| SLt | Sanli township (China) | 116.22 °E | 29.75 °N | (8/3) | 0.607 | 0.0111 | (5/1) | 0.000 | 0.0000 |
| NBp | Nanbei Port (China) | 116.17 °E | 29.72 °N | (8/1) | 0.000 | 0.0000 | (7/3) | 0.524 | 0.0023 |
| PYL | Poyang lake (China) | 116.43 °E | 28.87 °N | (7/1) | 0.000 | 0.0000 | (2/2) | 1.000 | 0.0041 |
| NCyl | Youlan, Nanchang (China) | 116.12 °E | 28.52 °N | (8/1) | 0.000 | 0.0000 | (3/1) | 0.000 | 0.0000 |
| NHL | Nanhu lake (China) | 114.03 °E | 30.02 °N | (7/1) | 0.000 | 0.0000 | (4/1) | 0.000 | 0.0000 |
| YNL | Yuni lake (China) | 112.20 °E | 30.00 °N | (8/1) | 0.000 | 0.0000 | (6/2) | 0.600 | 0.0024 |
| XT | Xiantao (China) | 113.40 °E | 30.30 °N | (8/1) | 0.000 | 0.0000 | (6/2) | 0.600 | 0.0024 |
| QJ | Qianjiang (China) | 112.60 °E | 30.40 °N | (7/1) | 0.000 | 0.0000 | (4/2) | 0.500 | 0.0182 |
| LZL | Liangzi lake (China) | 114.00 °E | 30.00 °N | (6/1) | 0.000 | 0.0000 | (2/2) | 1.000 | 0.0041 |
| HHL | Honghu lake (China) | 113.40 °E | 29.70 °N | (8/2) | 0.250 | 0.0003 | (2/2) | 1.000 | 0.0122 |
| CHL | Changhu lake (China) | 112.10 °E | 30.30 °N | (7/1) | 0.000 | 0.0000 | (6/3) | 0.733 | 0.0062 |
| YJ | Yuanjiang (China) | 112.37 °E | 28.85 °N | (7/1) | 0.000 | 0.0000 | (5/3) | 0.700 | 0.0089 |
| NX | Ningxiang (China) | 112.55 °E | 28.28 °N | (8/1) | 0.000 | 0.0000 | (4/3) | 0.833 | 0.0081 |
| DTL | Dongting lake (China) | 113.02 °E | 29.30 °N | (6/1) | 0.000 | 0.0000 | (2/2) | 1.000 | 0.0122 |
| DTLs | Dongting lakeside (China) | 113.13 °E | 29.35 °N | (8/2) | 0.250 | 0.0003 | (4/2) | 0.500 | 0.0061 |
| CQs | Chongqing suburb (China) | 106.53 °E | 29.55 °N | (7/1) | 0.000 | 0.0000 | (8/2) | 0.429 | 0.0017 |
| ZX | Zhongxian (China) | 108.03 °E | 30.28 °N | (6/2) | 0.533 | 0.0141 | (7/3) | 0.667 | 0.0058 |
| JY | Jianyang (China) | 104.55 °E | 30.38 °N | (6/2) | 0.333 | 0.0088 | (6/2) | 0.533 | 0.0022 |
| Sa | Saitama (Japan) | 139.65 °E | 35.85 °N | (18/3) | 0.569 | 0.0132 | (9/7) | 0.917 | 0.0180 |
| LA | Louisiana (USA) | 93.26 °W | 29.87 °N | (20/16) | 0.963 | 0.0086 | (9/4) | 0.778 | 0.0065 |

2.2. Variation and Genetic Structure
2.3. Scenario Testing


3. Discussion
4. Materials and Methods
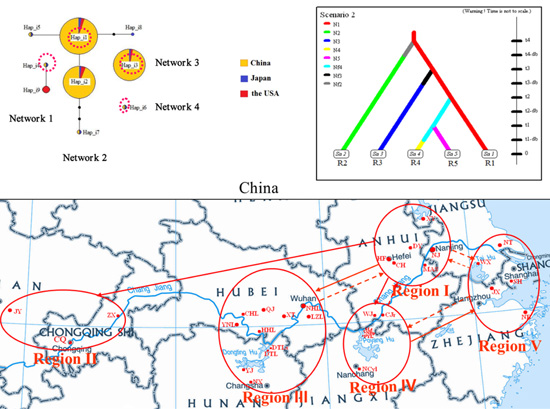
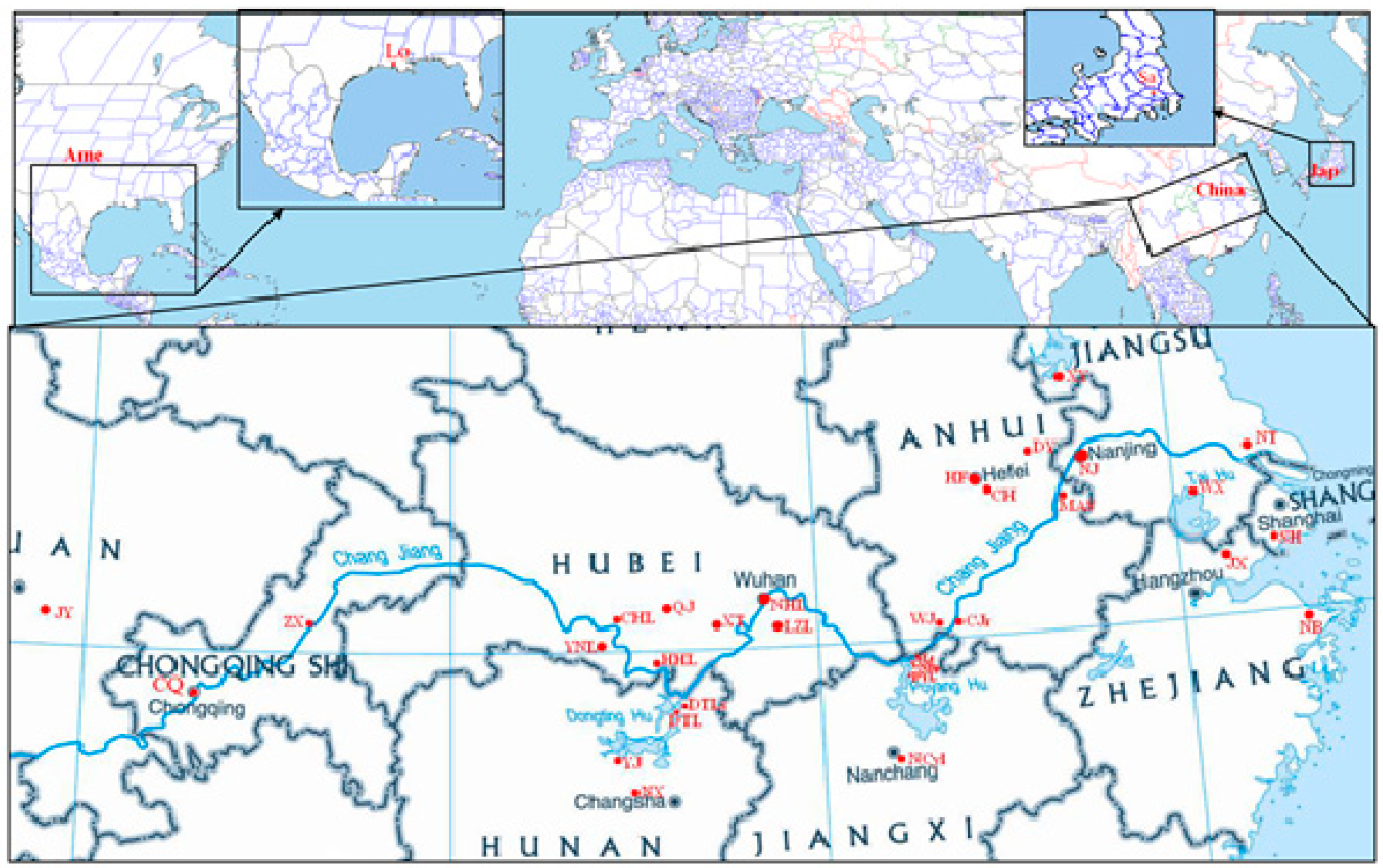
4.1. Sampling
4.2. DNA Isolation, Amplification and Sequencing
4.3. Data Analyses
4.3.1. Alignment of the Sequences
4.3.2. Variations and Genetic Diversity
4.3.3. Isolation by Distance (IBD)
4.3.4. Network Analysis
4.3.5. Scenario Testing
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pyšek, P.; Richardson, D.M. Invasive species, environmental change and management, and health. Annu. Rev. Environ. Resour. 2010, 35, 25–55. [Google Scholar] [CrossRef]
- Simberloff, D. Nonindigenous species: A global threat to biodiversity and stability. In Nature and Human Society: The Quest for a Sustainable World; Raven, P.W.T., Ed.; National Academy Press: Washington, DC, USA, 2000; pp. 325–336. [Google Scholar]
- Everett, R.A. Patterns and pathways of biological invasions. Trends Ecol. Evol. 2000, 15, 177–178. [Google Scholar] [CrossRef]
- Pimentel, D.; McNair, S.; Janecka, J.; Wightman, J.; Simmonds, C.; O’Connell, C.; Wong, E.; Russel, L.; Zern, J.; Aquino, T.; et al. Economic and environmental threats of alien plant, animal, and microbe invasions. Agric. Ecosyst. Environ. 2001, 84, 1–20. [Google Scholar] [CrossRef]
- Zhan, A.; Perepelizin, P.V.; Ghabooli, S.; Paolucci, E.; Sylvester, F.; Sardiña, P.; Cristescu, M.E.; MacIsaac, H.J. Scale-dependent post-establishment spread and genetic diversity in an invading mollusk in South America. Divers. Distrib. 2012, 18, 1042–1055. [Google Scholar] [CrossRef]
- Estoup, A.; Guillemaud, T. Reconstructing routes of invasion using genetic data: Why, how and so what? Mol. Ecol. 2010, 19, 4113–4130. [Google Scholar] [CrossRef] [PubMed]
- Guillemaud, T.; Beaumont, M.A.; Ciosi, M.; Cornuet, J.M.; Estoup, A. Inferring introduction routes of invasive species using approximate Bayesian computation on microsatellite data. Heredity 2010, 104, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Boissin, B.; Hurley, B.; Wingfield, M.J.; Vasaitis, R.; Stenlid, J.; Davis, C.; de Groot, P.; Ahumada, R.; Carnegie, A.; Goldarazena, A.; et al. Retracing the routes of introduction of invasive species: The case of the Sirex noctilio woodwasp. Mol. Ecol. 2012, 21, 5728–5744. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Guo, X.; Cao, X.; Deng, W.; Luo, W.; Wang, W.M. Population genetic structure and post-establishment dispersal patterns of the red swamp crayfish Procambarus clarkii in China. PLoS ONE 2012, 7, e40652. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Santos, F.; Beaumont, M.A.; Robert, C.P.; Marin, J.M.; Balding, D.J.; Guillemaud, T.; Estoup, A. Inferring population history with DIYABC: A user-friendly approach to approximate Bayesian computation. Bioinformatics 2008, 24, 2713–2719. [Google Scholar] [CrossRef] [PubMed]
- Cornuet, J.M.; Ravigne, V.; Estoup, A. Inference on population history and model checking using DNA sequence and microsatellite data with the software DIYABC (v1.0). BMC Bioinform. 2010, 11, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, N.; Estoup, A.; Toepfer, S.; Bourquet, D.; Lapchin, L.; Derridj, S.; Kim, K.S.; Reynaud, P.; Furlan, L.; Guillemaud, T. Multiple transatlantic introductions of the western corn rootworm. Science 2005, 310, 992. [Google Scholar] [CrossRef] [PubMed]
- Huner, J.V. Procambarus in North America and elsewhere. In Freshwater Crayfish: Biology, Management and Exploitation; Holdich, D.M., Lowery, R.S., Eds.; Croom Helm Ltd.: London, UK, 1988; pp. 239–261. [Google Scholar]
- Zhu, Z.Y.; Yue, G.H. Eleven polymorphic microsatellites isolated from red swamp crayfish, Procambarus clarkii. Mol. Ecol. Resour. 2008, 8, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Z.; Li, Z.; Liang, H.W.; Hu, G.F.; Wu, Q.C.; Zhou, G.W.; Luo, X.Z. Genetic diversity in four Procambarus clarkii populations in the lower reaches of the Yangtze River. Biodivers. Sci. 2009, 17, 518–523. (In Chinese) [Google Scholar]
- Yue, G.H.; Li, J.; Bai, Z.; Wang, C.M.; Feng, F. Genetic diversity and population structure of the invasive alien red swamp crayfish. Biol. Invasions 2010, 12, 2697–2706. [Google Scholar] [CrossRef]
- Siesa, M.E.; Manenti, R.; Padoa-Schioppa, E.; Bernardi, F.D.; Ficetola, G.F. Spatial autocorrelation and the analysis of invasion process from distribution data: A study with the crayfish Procambarus clarkii. Biol. Invasions 2011, 13, 2147–2160. [Google Scholar] [CrossRef]
- Li, S.; Xu, Y.; Du, L.; Yin, X.; Meng, X.; Xie, J. Investigation on and analysis of alien invasions in Chinese farming industry. Chin. Agric. Sci. Bull. 2005, 21, 156–159. [Google Scholar]
- Lavergne, S.; Molofsky, J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc. Natl. Acad. Soc. USA 2007, 104, 3883–3888. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.L.; Zhou, L.Z.; Zhang, B.W. Genetic patterns of invasive Procambarus clarkii population in the three river basins of Anhui Province. Biodivers. Sci. 2010, 18, 398–407. (In Chinese) [Google Scholar]
- Wenink, P.W.; Baker, A.J.; Tilanus, M.G.J. Mitochondrial control region sequences in two shorebird species: The turnstone and the dunlin, and their utility in population genetic studies. Mol. Biol. Evol. 1994, 11, 22–31. [Google Scholar] [PubMed]
- Palumbi, S.R. Nucleic acids II: The polymerase chain reaction. In Molecular Systematics; Hillis, D.M., Moritz, C., Mable, B.K., Eds.; Sinauer Associates, Inc.: Sunderland, MA, USA, 1996; pp. 205–247. [Google Scholar]
- Arias, A.; Freire, R.; Méndez, J.; Insua, A. Intron characterization and their potential as molecular markers for population studies in the scallops Aequipecten opercularis and Mimachlamys varia. Hereditas 2009, 146, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Palumbi, S.R.; Baker, C.S. Contrasting population structure from nuclear intron sequences and mtDNA of humpback whales. Mol. Biol. Evol. 1994, 11, 426–435. [Google Scholar] [PubMed]
- Castelloe, J.T.; Templeton, A.R. Root probabilities for intraspecific gene trees under neutral coalescent theory. Mol. Phylogenet. Evol. 1994, 3, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar] [PubMed]
- Xia, A.J.; Tang, J.Q. Status analysis and study idea of Procambarus clarkii. J. Aquac. 2006, 27, 9–11. (In Chinese) [Google Scholar]
- Dlugosch, K.M.; Parker, I.M. Founding events in species invasions: Genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 2008, 17, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Mayr, E. The nature of colonizations in birds. In The Genetics of Colonizing Species; Baker, H.G., Stebbins, G.L., Eds.; Academic Press: London, UK, 1965; pp. 29–43. [Google Scholar]
- Huey, R.B.; Gilchrist, G.W.; Carlson, M.L.; Berrigan, D.; Serra, L. Rapid evolution of a geographic cline in size in an introduced fly. Science 2000, 287, 308–309. [Google Scholar] [CrossRef] [PubMed]
- Reznick, D.N.; Ghalambor, C.K. The population ecology of contemporary adaptations: What empirical studies reveal about the conditions that promote adaptive evolution. Genetica 2001, 112, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Liu, Y.; Zhang, B. Invasion history of the oriental fruit fly, Bactrocera dorsalis, in the Pacific–Asia region: Two main invasion routes. PLoS ONE 2012, 7, e36176. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, L.A.; Mooney, H.A. Invasive alien species in an era of globalization. Front. Ecol. Environ. 2007, 4, 199–208. [Google Scholar] [CrossRef]
- Vitousek, P.M.; D’Antonio, C.M.; Loope, L.L.; Rejmanek, M.; Westbrooks, R. Introduced species: A significant component of human-caused global change. N. Z. J. Ecol. 1997, 21, 1–16. [Google Scholar]
- Li, Y.H.; Wang, W.M.; Liu, X.L.; Luo, W.; Zhang, J.; Gul, Y. DNA extraction from crayfish exoskeleton. Indian J. Exp. Biol. 2011, 49, 953–957. [Google Scholar] [PubMed]
- Li, Y.H.; Zheng, F.L.; Chen, H.Q.; Wang, H.Z.; Wang, L.Q.; Xu, D.P. Molecular cloning and sequence analysis of prophenoloxidase cDNA from haemocytes of the red swamp crayfish, Procambarus clarkii. Agric. Sci. China 2009, 8, 369–379. [Google Scholar] [CrossRef]
- Thompson, A.R.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The Clustal X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 24, 4876–4882. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Ne, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evol. Bioinform. 2005, 1, 47–50. [Google Scholar]
- Isolation by Distance. Available online: http://ibdws.sdsu.edu/~ibdws/ (accessed on 25 June 2015).
- Jensen, J.L.; Bohonak, A.J.; Kelley, S.T. Isolation by distance, web service. BMC Genet. 2005, 6. [Google Scholar] [CrossRef] [PubMed]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rate of base substitution through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Guo, X.; Chen, L.; Bai, X.; Wei, X.; Zhou, X.; Huang, S.; Wang, W. Inferring Invasion History of Red Swamp Crayfish (Procambarus clarkii) in China from Mitochondrial Control Region and Nuclear Intron Sequences. Int. J. Mol. Sci. 2015, 16, 14623-14639. https://doi.org/10.3390/ijms160714623
Li Y, Guo X, Chen L, Bai X, Wei X, Zhou X, Huang S, Wang W. Inferring Invasion History of Red Swamp Crayfish (Procambarus clarkii) in China from Mitochondrial Control Region and Nuclear Intron Sequences. International Journal of Molecular Sciences. 2015; 16(7):14623-14639. https://doi.org/10.3390/ijms160714623
Chicago/Turabian StyleLi, Yanhe, Xianwu Guo, Liping Chen, Xiaohui Bai, Xinlan Wei, Xiaoyun Zhou, Songqian Huang, and Weimin Wang. 2015. "Inferring Invasion History of Red Swamp Crayfish (Procambarus clarkii) in China from Mitochondrial Control Region and Nuclear Intron Sequences" International Journal of Molecular Sciences 16, no. 7: 14623-14639. https://doi.org/10.3390/ijms160714623
APA StyleLi, Y., Guo, X., Chen, L., Bai, X., Wei, X., Zhou, X., Huang, S., & Wang, W. (2015). Inferring Invasion History of Red Swamp Crayfish (Procambarus clarkii) in China from Mitochondrial Control Region and Nuclear Intron Sequences. International Journal of Molecular Sciences, 16(7), 14623-14639. https://doi.org/10.3390/ijms160714623