
Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System


Abstract
:1. Introduction
2. The Kynurenine Pathway
3. The Role of the KP in Immunoregulation
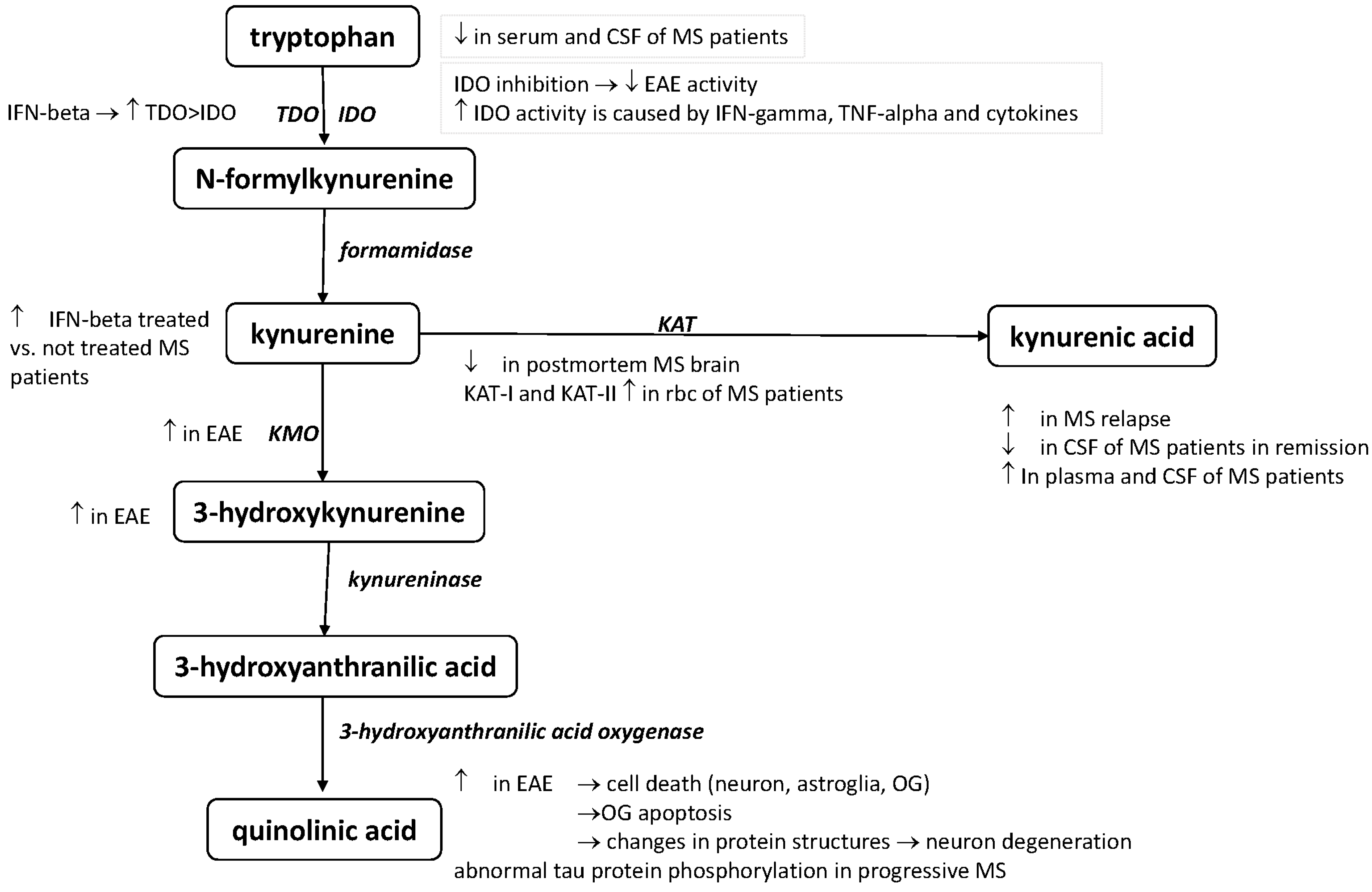
4. The Role of the KP in the Pathomechanism of MS
4.1. Preclinical Results
4.2. Clinical Results


{kind=link}
{kind=link}
| Alterations of the KP Metabolism | References |
|---|---|
| Elevated serum and CSF TRP levels in MS patients with acute relapse | [33] |
| Depressed serum and CSF TRP levels in MS | [34,35] |
| KAT I and KAT II serum levels were significantly higher in red blood cells of MS patients | [43] |
| Decrease the concentrations of KAT I and KAT II enzymes in postmortem MS brain sections | [45] |
| KYNA concentrations elevated in the plasma of MS patients | [43] |
| Elevated KYNA levels in the cerebrospinal fluid of MS patients | [44] |
| Low KYNA serum level in the CSF of MS patients in remission | [46] |
| Elevated KYNA level in the CSF with acute relapse | [47] |
| 3-HK is increased in EAE rats | [27] |
| KMO enzyme immunoreactivity has been found in cytoplasmic granules in the spinal cord and brainstem of rats with EAE | [27] |
| QUIN level increased in the spinal cords of EAE rats | [28] |
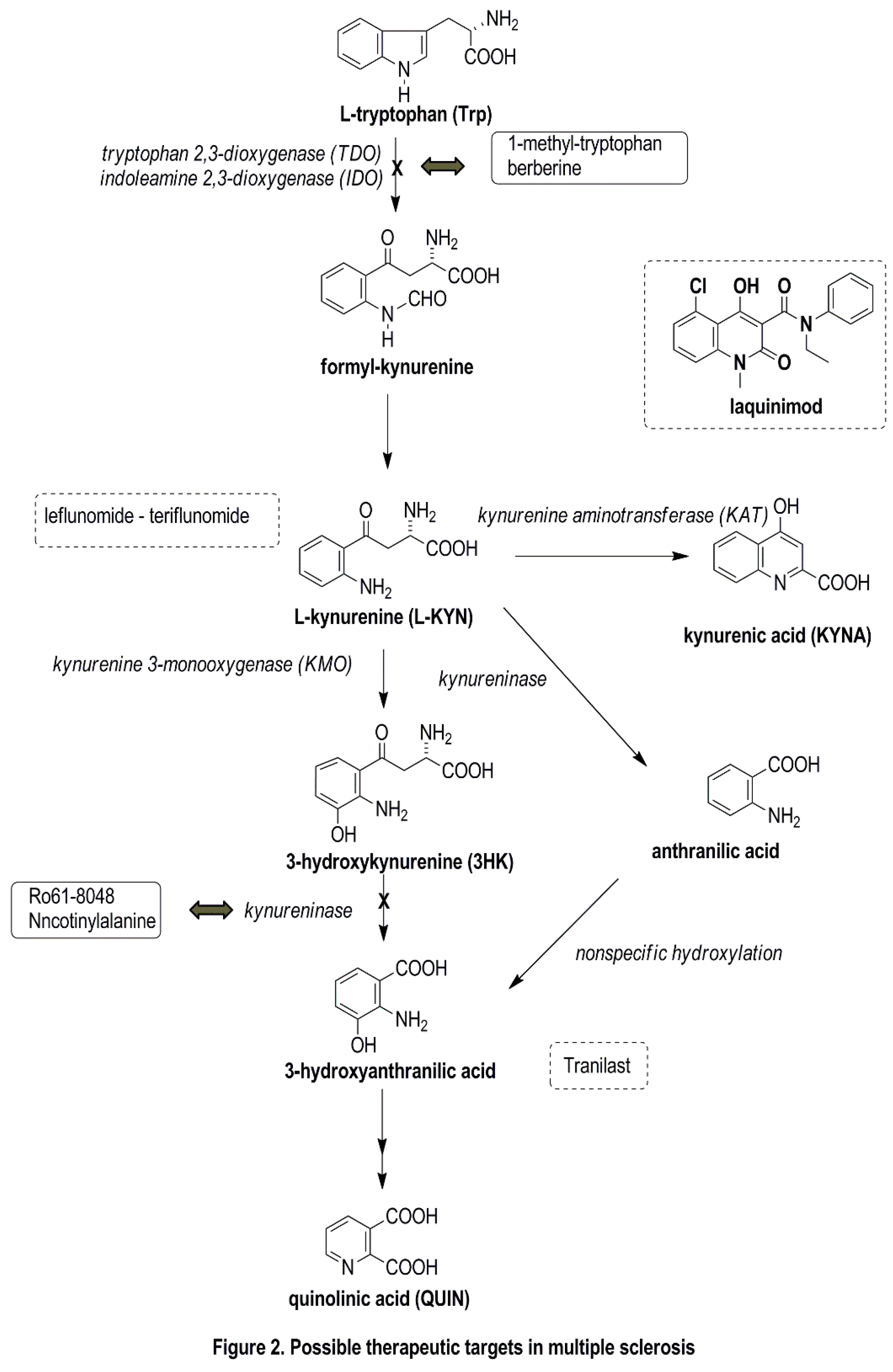
5. Possible Therapeutic Targets Related to the KP

6. Conclusions
Acknowledgments
Conflicts of Interest
List of Abbreviations
| 3HK | 3-hydroxi-kynurenine |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| CSF | cerebrospinal fluid |
| EAE | experimental autoimmune encephalomyelitis |
| IDO | indoleamine-2,3-dioxygenase |
| IFN-β | interferon-beta |
| IFN-γ | interferon-gamma |
| KAT | kynurenine-aminotransferase |
| KMO | kynurenine-3-monooxygenase |
| KP | kynurenine pathway |
| KYNA | kynurenic acid |
| L-KYN | L-kynurenine |
| MS | multiple sclerosis |
| MRI | magnetic resonance imaging |
| NMDA | N-methyl-d-aspartate |
| NO | nitrogen-monoxid |
| OG | oligodendrocyte |
| QUIN | quinolinic acid |
| RRMS | relapsing-remitting multiple sclerosis |
| TDO | tryptophane-2,3-dioxygenase |
| TNF-α | tumor necrosis factor-alpha |
| Trp | tryptophan |
References
- Lassmann, H.; Bruck, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- Nielsen, A.S.; Kinkel, R.P.; Madigan, N.; Tinelli, E.; Benner, T.; Mainero, C. Contribution of cortical lesion subtypes at 7T MRI to physical and cognitive performance in MS. Neurology 2013, 81, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.W.; Bo, L.; Mork, S.; Chang, A.; Trapp, B.D. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann. Neurol. 2001, 50, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L. Kynurenines in the Brain: From Experiments to Clinics; Nova Science Publishers: Hauppauge, NY, USA, 2005. [Google Scholar]
- Leklem, J.E. Quantitative aspects of tryptophan metabolism in humans and other species: A review. Am. J. Clin. Nutr. 1971, 24, 659–672. [Google Scholar] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell Mol. Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Terramani, T.; Lynch, G.; Baudry, M. A glycine site associated with N-methyl-d-aspartic acid receptors: Characterization and identification of a new class of antagonists. J. Neurochem. 1989, 52, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993, 45, 309–379. [Google Scholar] [PubMed]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rozsa, E.; Robotka, H.; Vecsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural. Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits α7 nicotinic receptor activity and increases non-α7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [PubMed]
- Marchi, M.; Risso, F.; Viola, C.; Cavazzani, P.; Raiteri, M. Direct evidence that release-stimulating α7 nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J. Neurochem. 2002, 80, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W.; Perkins, M.N. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Perez-De La Cruz, V.; Carrillo-Mora, P.; Santamaria, A. Quinolinic acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, 1–8. [Google Scholar] [PubMed]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Mandi, Y.; Vecsei, L. The kynurenine system and immunoregulation. J. Neural. Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Belladonna, M.L.; Puccetti, P.; Orabona, C.; Fallarino, F.; Vacca, C.; Volpi, C.; Gizzi, S.; Pallotta, M.T.; Fioretti, M.C.; Grohmann, U. Immunosuppression via tryptophan catabolism: The role of kynurenine pathway enzymes. Transplantation 2007, 84, S17–S20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.; Varo, N.; Alegre, E.; Diaz, A.; Melero, I. Immunosuppression routed via the kynurenine pathway: A biochemical and pathophysiologic approach. Adv. Clin. Chem. 2008, 45, 155–197. [Google Scholar] [PubMed]
- Paterson, P.Y. Autoimmune diseases of myelin. Prog. Clin. Biol. Res. 1980, 49, 19–36. [Google Scholar] [PubMed]
- Gold, R.; Linington, C.; Lassmann, H. Understanding pathogenesis and therapy of multiple sclerosis via animal models: 70 years of merits and culprits in experimental autoimmune encephalomyelitis research. Brain 2006, 129, 1953–1971. [Google Scholar] [CrossRef] [PubMed]
- Kwidzinski, E.; Bunse, J.; Aktas, O.; Richter, D.; Mutlu, L.; Zipp, F.; Nitsch, R.; Bechmann, I. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 2005, 19, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Cozzi, A.; Ballerini, C.; Massacesi, L.; Moroni, F. Kynurenine 3-mono-oxygenase activity and neurotoxic kynurenine metabolites increase in the spinal cord of rats with experimental allergic encephalomyelitis. Neuroscience 2001, 102, 687–695. [Google Scholar] [CrossRef]
- Flanagan, E.M.; Erickson, J.B.; Viveros, O.H.; Chang, S.Y.; Reinhard, J.F., Jr. Neurotoxin quinolinic acid is selectively elevated in spinal cords of rats with experimental allergic encephalomyelitis. J. Neurochem. 1995, 64, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Cammer, W. Oligodendrocyte killing by quinolinic acid in vitro. Brain Res. 2001, 896, 157–160. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Wang, L.; Brew, B.J. Quinolinic acid selectively induces apoptosis of human astrocytes: Potential role in AIDS dementia complex. J. Neuroinflamm. 2005, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, S.J.; Armati, P.J.; Guillemin, G.J.; Brew, B.J. Chronic exposure of human neurons to quinolinic acid results in neuronal changes consistent with aids dementia complex. AIDS 1998, 12, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Pierozan, P.; Zamoner, A.; Soska, A.K.; Silvestrin, R.B.; Loureiro, S.O.; Heimfarth, L.; Mello e Souza, T.; Wajner, M.; Pessoa-Pureur, R. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp. Neurol. 2010, 224, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Monaco, F.; Fumero, S.; Mondino, A.; Mutani, R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J Neurol. Neurosurg. Psychiatry 1979, 42, 640–641. [Google Scholar] [CrossRef] [PubMed]
- Rudzite, V.; Berzinsh, J.; Grivane, I.; Fuchs, D.; Baier-Bitterlich, G.; Wachter, H. Serum tryptophan, kynurenine, and neopterin in patients with Guillain-Barre-syndrome (GBS) and multiple sclerosis (MS). Adv. Exp. Med. Biol. 1996, 398, 183–187. [Google Scholar] [PubMed]
- Sandyk, R. Tryptophan availability and the susceptibility to stress in multiple sclerosis: A hypothesis. Int. J. Neurosci. 1996, 86, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sadowska-Bartosz, I.; Adamczyk-Sowa, M.; Gajewska, A.; Bartosz, G. Oxidative modification of blood serum proteins in multiple sclerosis after interferon or mitoxantrone treatment. J. Neuroimmunol. 2014, 266, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.M.; Hampton, D.W.; Patani, R.; Pryce, G.; Crowther, R.A.; Reynolds, R.; Franklin, R.J.; Giovannoni, G.; Compston, D.A.; Baker, D.; et al. Abnormally phosphorylated tau is associated with neuronal and axonal loss in experimental autoimmune encephalomyelitis and multiple sclerosis. Brain 2008, 131, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.; Rondot, P.; Catinot, L.; Falcoff, E.; Kirchner, H.; Wietzerbin, J. Increased production of interferon γ and tumor necrosis factor precedes clinical manifestation in multiple sclerosis: Do cytokines trigger off exacerbations? Acta Neurol. Scand. 1988, 78, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, L.A.; Kerr, S.J.; Smythe, G.; Brew, B.J. Quinolinic acid production by macrophages stimulated with IFN-γ, TNF-α, and IFN-α. J. Interferon Cytokine Res. 1997, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Guillemin, G.J.; Pemberton, L.; Kerr, S.; Nath, A.; Smythe, G.A.; Brew, B.J. Quinolinic acid is produced by macrophages stimulated by platelet activating factor, nef and tat. J. Neurovirol. 2001, 7, 56–60. [Google Scholar] [PubMed]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar] [PubMed]
- Guillemin, G.J.; Kerr, S.J.; Pemberton, L.A.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, B.J. IFN-β1b induces kynurenine pathway metabolism in human macrophages: Potential implications for multiple sclerosis treatment. J. Interferon Cytokine Res. 2001, 21, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vecsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Kepplinger, B.; Baran, H.; Kainz, A.; Ferraz-Leite, H.; Newcombe, J.; Kalina, P. Age-related increase of kynurenic acid in human cerebrospinal fluid—IgG and β2-microglobulin changes. Neuro Signals 2005, 14, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Brew, B.J.; Sundaram, G.; Guillemin, G.J. Understanding the roles of the kynurenine pathway in multiple sclerosis progression. Int. J. Tryptophan Res. 2010, 3, 157–167. [Google Scholar] [PubMed]
- Rejdak, K.; Bartosik-Psujek, H.; Dobosz, B.; Kocki, T.; Grieb, P.; Giovannoni, G.; Turski, W.A.; Stelmasiak, Z. Decreased level of kynurenic acid in cerebrospinal fluid of relapsing-onset multiple sclerosis patients. Neurosci. Lett. 2002, 331, 63–65. [Google Scholar] [CrossRef]
- Rejdak, K.; Petzold, A.; Kocki, T.; Kurzepa, J.; Grieb, P.; Turski, W.A.; Stelmasiak, Z. Astrocytic activation in relation to inflammatory markers during clinical exacerbation of relapsing-remitting multiple sclerosis. J. Neural. Transm. 2007, 114, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Paul, C.; Bolton, C. Modulation of blood-brain barrier dysfunction and neurological deficits during acute experimental allergic encephalomyelitis by the N-methyl-d-aspartate receptor antagonist memantine. J. Pharmacol. Exp. Ther. 2002, 302, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Kynurenic acid antagonists and kynurenine pathway inhibitors. Exp. Opin. Investig. Drugs 2001, 10, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Carpenedo, R.; Molina, M.T.; Mattoli, L.; Pellicciari, R.; Moroni, F. Comparison of the neurochemical and behavioral effects resulting from the inhibition of kynurenine hydroxylase and/or kynureninase. J. Neurochem. 1995, 65, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Colabroy, K.L.; Zhai, H.; Li, T.; Ge, Y.; Zhang, Y.; Liu, A.; Ealick, S.E.; McLafferty, F.W.; Begley, T.P. The mechanism of inactivation of 3-hydroxyanthranilate-3,4-dioxygenase by 4-chloro-3-hydroxyanthranilate. Biochemistry 2005, 44, 7623–7631. [Google Scholar] [CrossRef] [PubMed]
- Walsh, H.A.; O'Shea, K.C.; Botting, N.P. Comparative inhibition by substrate analogues 3-methoxy- and 3-hydroxydesaminokynurenine and an improved 3 step purification of recombinant human kynureninase. BMC Biochem. 2003, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platten, M.; Ho, P.P.; Youssef, S.; Fontoura, P.; Garren, H.; Hur, E.M.; Gupta, R.; Lee, L.Y.; Kidd, B.A.; Robinson, W.H.; et al. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science 2005, 310, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Bruck, W.; Wegner, C. Insight into the mechanism of laquinimod action. J. Neurol. Sci. 2011, 306, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O. Oral laquinimod treatment in multiple sclerosis. Neurologia 2011, 26, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.; Barkhof, F.; Sandberg-Wollheim, M.; Linde, A.; Nordle, O.; Nederman, T. Treatment with laquinimod reduces development of active MRI lesions in relapsing MS. Neurology 2005, 64, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, M.; Gritzman, T.; Orbach, R.; Tuller, T.; Feldman, A.; Achiron, A. Laquinimod suppress antigen presentation in relapsing-remitting multiple sclerosis: In-vitro high-throughput gene expression study. J. Neuroimmunol. 2010, 221, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.P.; Abbas, N.; Volkmann, I.; Nennesmo, I.; Levi, M.; Wahren, B.; Winblad, B.; Hedlund, G.; Zhu, J. Suppression of experimental autoimmune neuritis by ABR-215062 is associated with altered Th1/Th2 balance and inhibited migration of inflammatory cells into the peripheral nerve tissue. Neuropharmacology 2002, 42, 731–739. [Google Scholar] [CrossRef]
- Wegner, C.; Stadelmann, C.; Pfortner, R.; Raymond, E.; Feigelson, S.; Alon, R.; Timan, B.; Hayardeny, L.; Bruck, W. Laquinimod interferes with migratory capacity of T cells and reduces IL-17 levels, inflammatory demyelination and acute axonal damage in mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2010, 227, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, R.; Saada, R.; Eilam, R.; Hayardeny, L.; Sela, M.; Arnon, R. Oral treatment with laquinimod augments regulatory T-cells and brain-derived neurotrophic factor expression and reduces injury in the CNS of mice with experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2012, 251, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Topphoff, U.; Shetty, A.; Varrin-Doyer, M.; Molnarfi, N.; Sagan, S.A.; Sobel, R.A.; Nelson, P.A.; Zamvil, S.S. Laquinimod, a quinoline-3-carboxamide, induces type II myeloid cells that modulate central nervous system autoimmunity. PLoS ONE 2012, 7, e33797. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Prod'homme, T.; Youssef, S.; Dunn, S.E.; Rundle, C.D.; Lee, L.; Patarroyo, J.C.; Stuve, O.; Sobel, R.A.; Steinman, L.; et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat. Med. 2007, 13, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Eng. J. Med. 2012, 366, 1000–1009. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, G.; Brew, B.J.; Jones, S.P.; Adams, S.; Lim, C.K.; Guillemin, G.J. Quinolinic acid toxicity on oligodendroglial cells: Relevance for multiple sclerosis and therapeutic strategies. J. Neuroinflamm. 2014, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharmacol. 2012, 81, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Volpi, C.; Fazio, F.; Notartomaso, S.; Vacca, C.; Busceti, C.; Bicciato, S.; Battaglia, G.; Bruno, V.; Puccetti, P.; et al. Metabotropic glutamate receptor-4 modulates adaptive immunity and restrains neuroinflammation. Nat. Med. 2010, 16, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Zappulla, C.; Notartomaso, S.; Busceti, C.; Bessede, A.; Scarselli, P.; Vacca, C.; Gargaro, M.; Volpi, C.; Allegrucci, M.; et al. Cinnabarinic acid, an endogenous agonist of type-4 metabotropic glutamate receptor, suppresses experimental autoimmune encephalomyelitis in mice. Neuropharmacology 2014, 81, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Rodriguez, M. Multiple sclerosis, seizures, and antiepileptics: Role of IL-18, IDO, and melatonin. Eur. J. Neurol. 2011, 18, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Gonsette, R.E. Endogenous neuroprotection in multiple sclerosis. Acta Neurol. Belg. 2010, 110, 26–35. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajda, C.; Majláth, Z.; Pukoli, D.; Vécsei, L. Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System. Int. J. Mol. Sci. 2015, 16, 18270-18282. https://doi.org/10.3390/ijms160818270
Rajda C, Majláth Z, Pukoli D, Vécsei L. Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System. International Journal of Molecular Sciences. 2015; 16(8):18270-18282. https://doi.org/10.3390/ijms160818270
Chicago/Turabian StyleRajda, Cecilia, Zsófia Majláth, Dániel Pukoli, and László Vécsei. 2015. "Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System" International Journal of Molecular Sciences 16, no. 8: 18270-18282. https://doi.org/10.3390/ijms160818270
APA StyleRajda, C., Majláth, Z., Pukoli, D., & Vécsei, L. (2015). Kynurenines and Multiple Sclerosis: The Dialogue between the Immune System and the Central Nervous System. International Journal of Molecular Sciences, 16(8), 18270-18282. https://doi.org/10.3390/ijms160818270