
Research Advances on Pathways of Nickel-Induced Apoptosis

Abstract
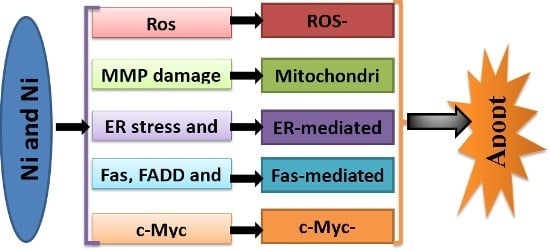
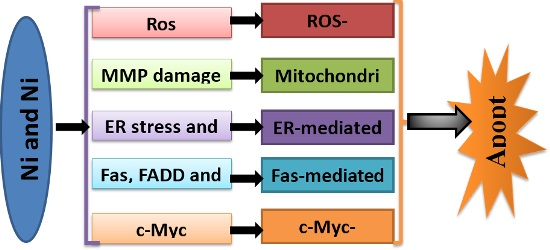
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Chemical Properties and Toxicity of Nickel
1.1. Chemical Properties of Ni
1.2. Biological Properties of Ni
1.3. Ni Toxicity
2. Biological Characteristics of Cell Apoptosis
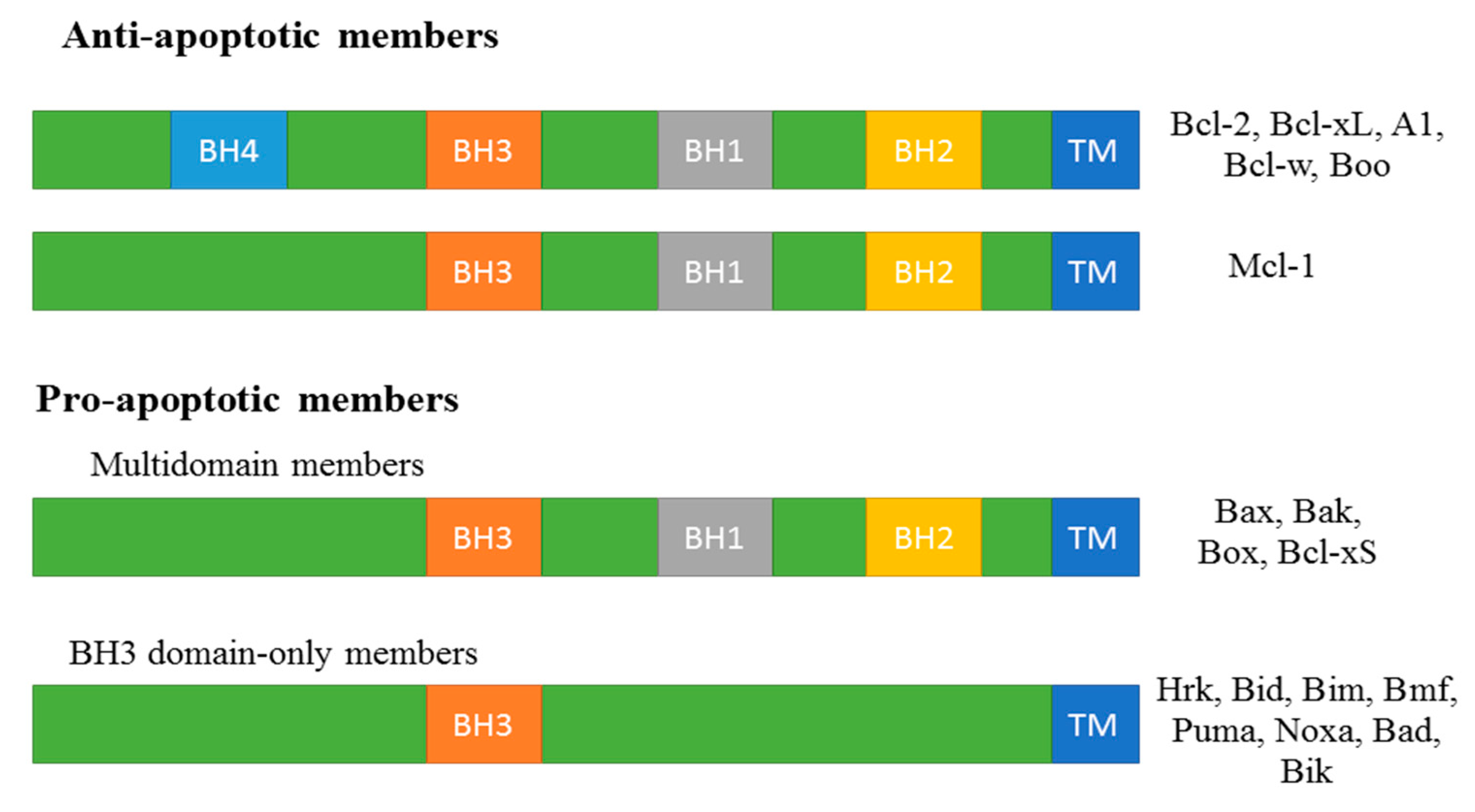
2.1. Bcl-2 Family Protein in Apoptosis


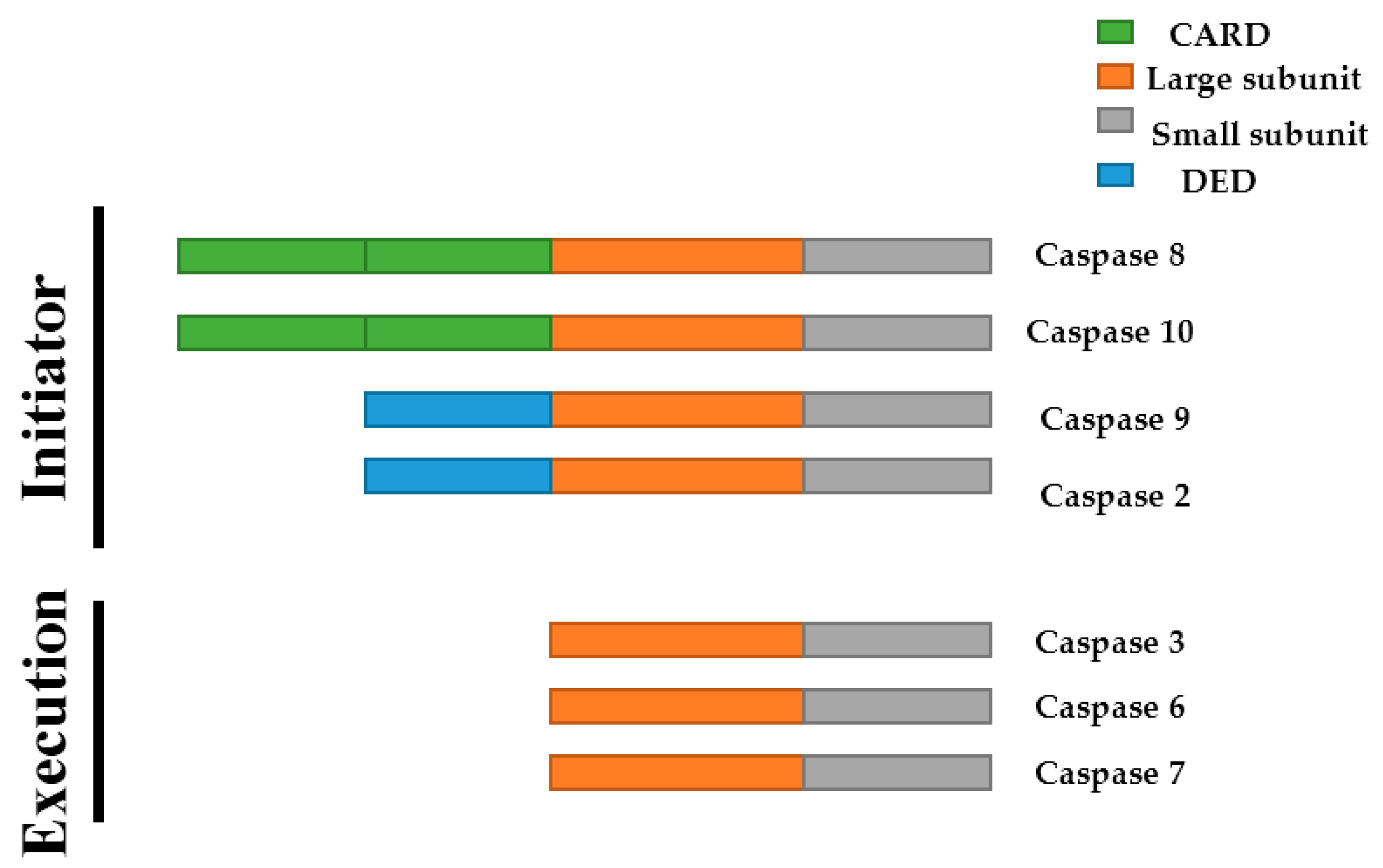
2.2. Caspases in Apoptosis

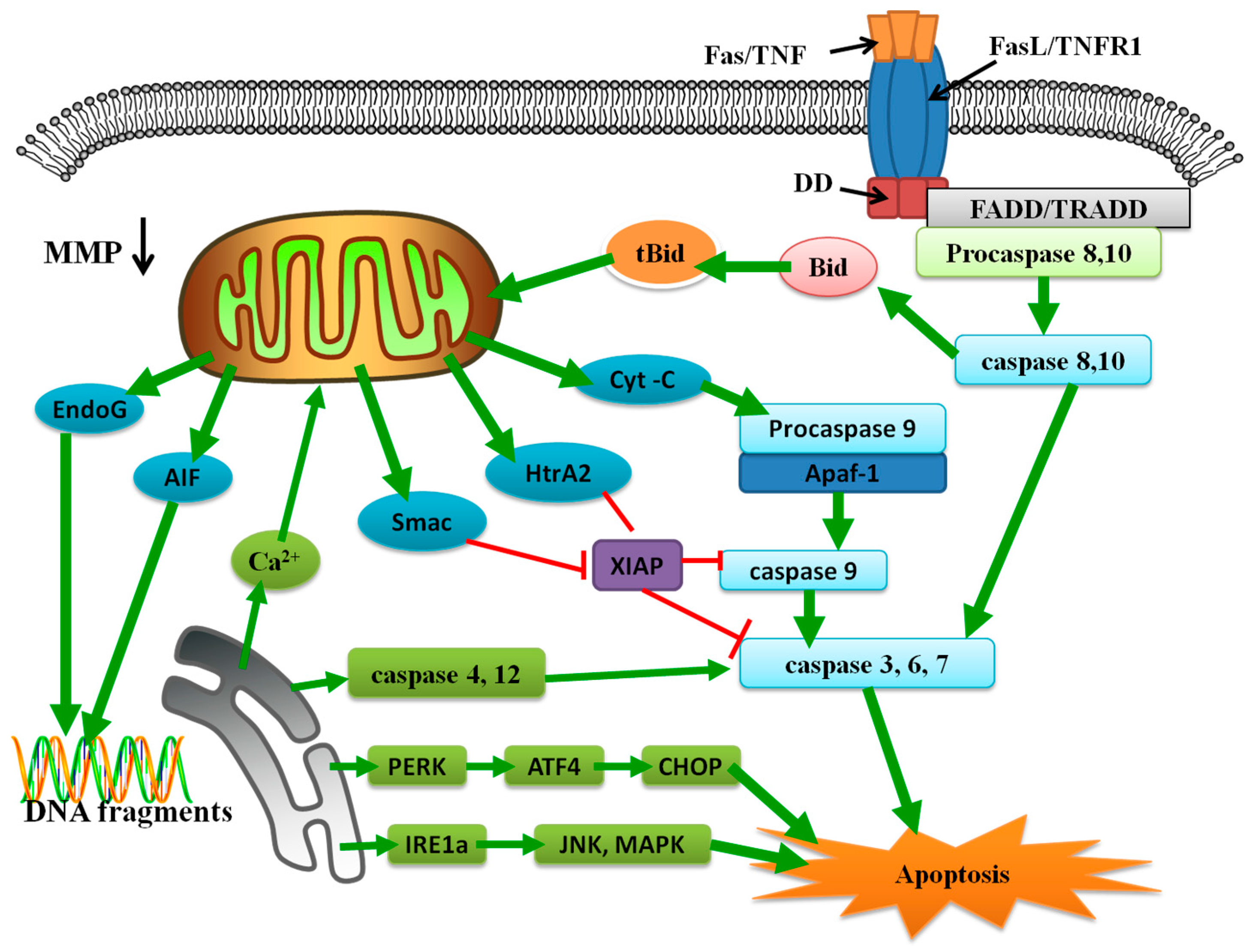
2.3. Extrinsic Pathway in Apoptosis
2.4. Intrinsic Pathway in Apoptosis
2.4.1. Mitochondria in the Intrinsic Pathway
2.4.2. ER in the Intrinsic Pathway
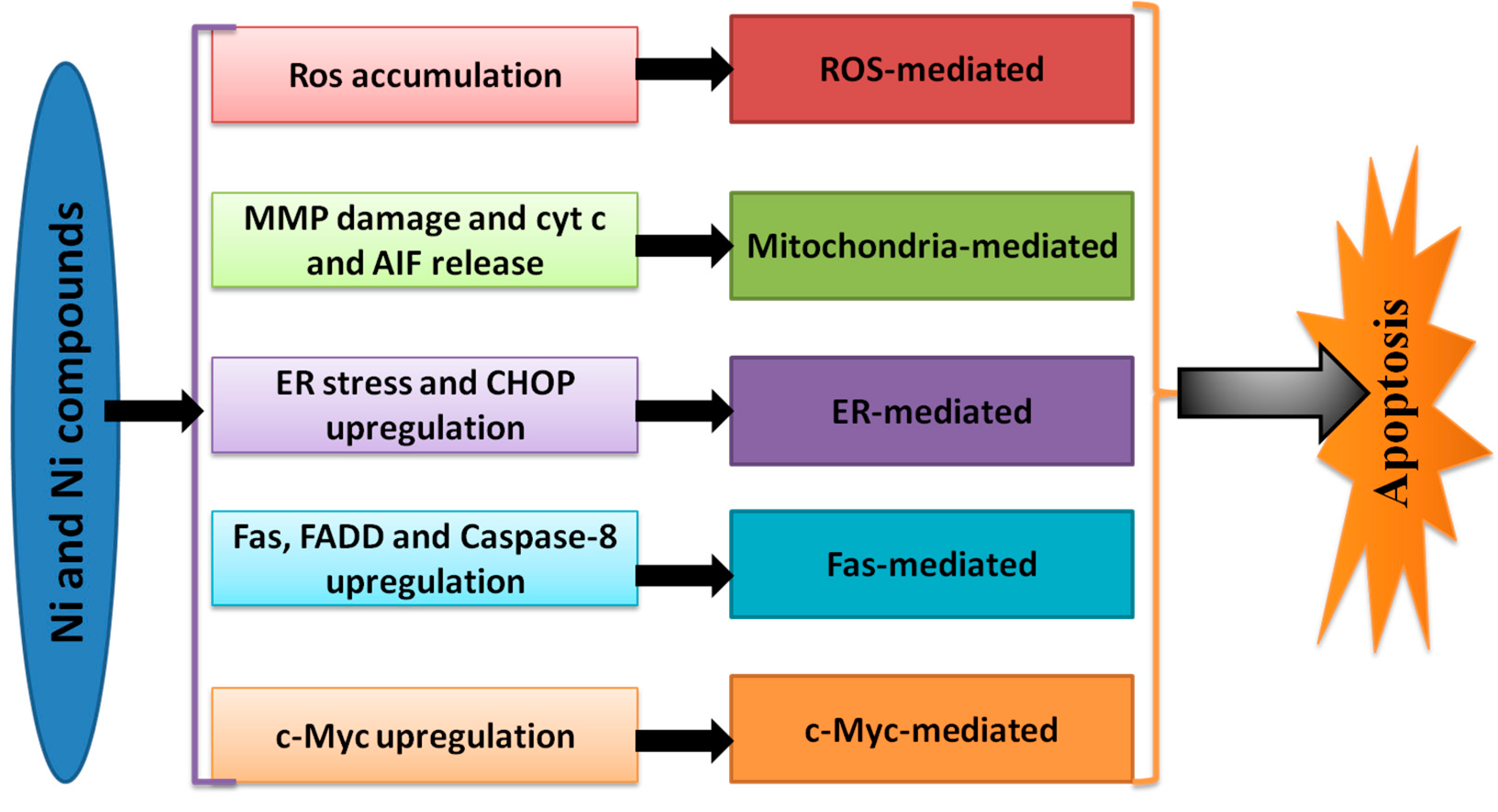
3. Apoptosis Induced by Ni
4. Pathways or Mechanisms of Ni-Induced Cell Apoptosis
4.1. ROS-Mediated Apoptosis
4.2. The Extrinsic Pathway of Ni-Induced Apoptosis
4.3. The Intrinsic Pathway of Ni-Induced Apoptosis
4.4. Others
5. Conclusions and Future Perspectives

Acknowledgments
Author Contributions
Conflicts of Interest
References
- Greenwood, N.N.; Earnshaw, A.; Earnshaw, A. Chemistry of the Elements; Butterworth-Heinemann: Oxford, UK, 1997. [Google Scholar]
- Doll, R. Nickel exposure: A human health hazard. IARC Sci. Publ. 1984, 53, 3–21. [Google Scholar] [PubMed]
- Gurley, L.R.; Tobey, R.A.; Valdez, J.G.; Halleck, M.S.; Barham, S.S. Biological availability of nickel arsenides: Toxic effects of particulate Ni5As2. Sci. Total Environ. 1983, 28, 415–432. [Google Scholar] [CrossRef]
- Rando, D.; Steglitz, U.; Morsdorf, G.; Kaltwasser, H. Nickel availability and urease expression in Proteus mirabilis. Arch. Microbiol. 1990, 154, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Schaumlöffel, D. Nickel species: Analysis and toxic effects. J. Trace Elem. Med. Biol. 2012, 26, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A.; Costa, M. Elucidating the mechanisms of nickel compound uptake: A review of particulate and nano-nickel endocytosis and toxicity. Toxicol. Appl. Pharmacol. 2012, 260, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, J.; Minocha, J.; Laumann, A. Body piercing: Complications and prevention of health risks. Am. J. Clin. Dermatol. 2012, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.G.; Durando, J.; Oller, A.R.; Merkel, D.J.; Marone, P.A.; Bates, H.K. Acute oral toxicity of nickel compounds. Regul. Toxicol. Pharm. 2012, 62, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Cempel, M.; Nikel, G. Nickel: A review of its sources and environmental toxicology. Pol. J. Environ. Stud. 2006, 15, 375–382. [Google Scholar]
- Grandjean, P. Human exposure to nickel. IARC Sci. Publ. 1984, 469–485. [Google Scholar]
- Tyagi, R.; Rana, P.; Gupta, M.; Khan, A.R.; Bhatnagar, D.; Bhalla, P.J.; Chaturvedi, S.; Tripathi, R.P.; Khushu, S. Differential biochemical response of rat kidney towards low and high doses of NiCl2 as revealed by NMR spectroscopy. J. Appl. Toxicol. 2013, 33, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Leach, R. Studies on nickel metabolism: Interaction with other mineral elements. Poultry Sci. 1979, 58, 591–596. [Google Scholar] [CrossRef]
- Wen, Z.T.; Burne, R.A. Functional genomics approach to identifying genes required for biofilm development by Streptococcus mutans. Appl. Environ. Microbiol. 2002, 68, 1196–1203. [Google Scholar] [CrossRef] [PubMed]
- Kirchgessner, M.; Perth, J.; Schnegg, A. Deficient nickel supply and the content of calcium, magnesium and phosphorus inthe bone of growing rats. Arch. Tierernahr. 1980, 30, 805–810. [Google Scholar] [CrossRef] [PubMed]
- Poonkothai, M.V.B.S. Nickel as an essential element and a toxicant. Int. J. Environ. Sci. 2012, 1, 285–288. [Google Scholar]
- Coogan, T.P.; Latta, D.M.; Imbra, R.J.; Costa, M. Effect of nickel(II) on DNA-protein interactions. Biol. Trace Elem. Res. 1989, 21, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Schnegg, A.; Kirchgessner, M. Absorption and metabolic efficiency of iron in nickel deficiency. Int. J. Vitam. Nutr. Res. 1976, 46, 96–99. [Google Scholar] [PubMed]
- Tallkvist, J.; Tjalve, H. Effect of dietary iron-deficiency on the disposition of nickel in rats. Toxicol. Lett. 1997, 92, 131–138. [Google Scholar] [CrossRef]
- Haber, L.; Erdreicht, L.; Diamond, G.; Maier, A.; Ratney, R.; Zhao, Q.; Dourson, M. Hazard identification and dose response of inhaled nickel-soluble salts. Regul. Toxicol. Pharmacol. 2000, 31, 210–230. [Google Scholar] [CrossRef] [PubMed]
- Oller, A.R.; Costa, M.; Oberdorster, G. Carcinogenicity assessment of selected nickel compounds. Toxicol. Appl. Pharmacol. 1997, 143, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Wataha, J.C.; Lockwood, P.E.; Schedle, A.; Noda, M.; Bouillaguet, S. Ag, Cu, Hg and Ni ions alter the metabolism of human monocytes during extended low-dose exposures. J. Oral Rehabil. 2002, 29, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Das, S.N.; Dhundasi, S.A. Nickel, its adverse health effects & oxidative stress. Indian J. Med. Res. 2008, 128, 412–425. [Google Scholar] [PubMed]
- Li, X.; Zhong, F. Nickel induces interleukin-1β secretion via the NLRP3-ASC-caspase-1 pathway. Inflammation 2014, 37, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; He, M.; Zhong, M.; Li, L.; Lu, Y.; Zhang, Y.; Zhang, L.; Yu, Z.; Zhou, Z. The neuroprotective effects of taurine against nickel by reducing oxidative stress and maintaining mitochondrial function in cortical neurons. Neurosci. Lett. 2015, 590, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Gathwan, K.H.; Al-Karkhi, I.H.T.; Jaffar Al-Mulla, E.A. Hepatic toxicity of nickel chloride in mice. Res. Chem. Intermed. 2012, 39, 2537–2542. [Google Scholar] [CrossRef]
- Scutariu, M.D.; Ciupilan, C. Nickel and magnesium effects in the rat kidney, treated with acid retinoic. Comparative study. Rev. Med. Chir. Soc. Med. Nat. Iasi. 2007, 111, 744–747. [Google Scholar] [PubMed]
- Chen, C.Y.; Lin, T.K.; Chang, Y.C.; Wang, Y.F.; Shyu, H.W.; Lin, K.H.; Chou, M.C. Nickel (II)-induced oxidative stress, apoptosis, G2/M arrest, and genotoxicity in normal rat kidney cells. J. Toxicol. Environ. Health A 2010, 73, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Goodman, J.E.; Prueitt, R.L.; Dodge, D.G.; Thakali, S. Carcinogenicity assessment of water-soluble nickel compounds. Crit. Rev. Toxicol. 2009, 39, 365–417. [Google Scholar] [CrossRef] [PubMed]
- Forgacs, Z.; Massanyi, P.; Lukac, N.; Somosy, Z. Reproductive toxicology of nickel—Review. J. Environ. Sci. Health A 2012, 47, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Duman, F.; Ozturk, F. Nickel accumulation and its effect on biomass, protein content and antioxidative enzymes in roots and leaves of watercress (Nasturtium officinale R. Br.). J. Environ. Sci. 2010, 22, 526–532. [Google Scholar] [CrossRef]
- Kasprzak, K.S.; Sunderman, F.W., Jr.; Salnikow, K. Nickel carcinogenesis. Mutat. Res. 2003, 533, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Spears, J.W.; Harvey, R.W.; Samsell, L.J. Effects of dietary nickel and protein on growth, nitrogen metabolism and tissue concentrations of nickel, iron, zinc, manganese and copper in calves. J. Nutr. 1986, 116, 1873–1882. [Google Scholar] [PubMed]
- Das, K.K.; Dasgupta, S. Effect of nickel on testicular nucleic acid concentrations of rats on protein restriction. Biol. Trace Elem. Res. 2000, 73, 175–180. [Google Scholar] [CrossRef]
- Ragsdale, S.W. Nickel biochemistry. Curr. Opin. Chem. Biol. 1998, 2, 208–215. [Google Scholar] [CrossRef]
- Denkhausa, E.S.K. Nickel essentiality, toxicity, and carcinogenicity. Crit. Rev. Oncol. Hemat. 2002, 42, 35–36. [Google Scholar] [CrossRef]
- Krockova, J.Z.; Massanyi, P.; Sirotkin, A.V.; Pivko, J.; Makarevich, A.V.; Lukac, N.; Capcarova, M.; Toman, R.; Polakova, Z. Nickel induced structural and functional alterations in mouse Leydig cells in vitro. J. Trace Elem. Med. Biol. 2011, 25, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z. Nickel carbonyl: Toxicity and human health. Sci. Total Environ. 1994, 148, 293–298. [Google Scholar] [CrossRef]
- Das, K.K.; Buchner, V. Effect of nickel exposure on peripheral tissues: Role of oxidative stress in toxicity and possible protection by ascorbic acid. Rev. Environ. Health 2007, 22, 157–173. [Google Scholar] [CrossRef] [PubMed]
- IARC. IARC Monograph on the Evaluation of Carcinogenic Risks to Humans; International Agency for Research on Cancer: Lyon, France, 1990; Volume 49, pp. 318–411. [Google Scholar]
- Cavallo, D.; Ursini, C.; Setini, A.; Chianese, C.; Piegari, P.; Perniconi, B.; Iavicoli, S. Evaluation of oxidative damage and inhibition of DNA repair in an in vitro study of nickel exposure. Toxicol. Vitro 2003, 17, 603–607. [Google Scholar] [CrossRef]
- Salnikow, K.; Zhitkovich, A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: Nickel, arsenic, and chromium. Chem. Res. Toxicol. 2008, 21, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ren, T.; Xiao, C.; Li, H.; Wu, T. Nickel promotes the invasive potential of human lung cancer cells via TLR4/MyD88 signaling. Toxicology 2011, 285, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol. 2008, 82, 493–512. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.K.; Bai, C.; Subramanian, K.S. DNA-protein crosslinks induced by nickel compounds in isolated rat lymphocytes: Role of reactive oxygen species and specific amino acids. Toxicol. Appl. Pharmacol. 2001, 170, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.X.; Chen, J.K.; Wu, Z.L. Detection of DNA strand breaks, DNA-protein crosslinks, and telomerase activity in nickel-transformed BALB/c-3T3 cells. Teratog. Carcinog. Mutagen. 2001, 21, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.W.; Reid, B.L. Nickel toxicity in growing chicks. J. Nutr. 1968, 95, 612–616. [Google Scholar] [PubMed]
- Szilagyi, M.; Szentmihalyi, S.; Anke, M. Changes in Some of the Biochemical Parameters in Ni and Mo Deficient Animals (Goat, Sheep, Pig, Chicken, Rat). Available online: http://agris.fao.org/agris-search/search.do?recordID=HU8200908 (accessed on 9 April 2015).
- Amudha, K.; Pari, L. Beneficial role of naringin, a flavanoid on nickel induced nephrotoxicity in rats. Chem. Biol. Interact. 2011, 193, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Guo, H.; Deng, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Wang, X.; Wu, B.; Li, J.; et al. Inhibitive Effects of Nickel Chloride (NiCl2) on Thymocytes. Biol. Trace Elem. Res. 2015, 164, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Li, J.; Yin, S.; Guo, H.; Deng, J.; Cui, H. Effects of nickel chloride on histopathological lesions and oxidative damage in the thymus. Health 2014, 6, 2875. [Google Scholar] [CrossRef]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B. Effect of dietary nickel chloride on splenic immune function in broilers. Biol. Trace Elem. Res. 2014, 159, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B. Downregulation of TLR4 and 7 mRNA expression levels in broiler’s spleen caused by diets supplemented with nickel chloride. Biol. Trace Elem. Res. 2014, 158, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Deng, J.; Yin, S.; et al. NiCl2-down-regulated antioxidant enzyme mRNA expression causes oxidative damage in the broiler’s kidney. Biol. Trace Elem. Res. 2014, 162, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride restrains the development of small intestine in broilers. Biol. Trace Elem. Res. 2013, 155, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B.; Guo, H. Toxic effect of NiCl2 on development of the bursa of Fabricius in broiler chickens. Oncotarget 2015. [Google Scholar] [CrossRef]
- Guo, H.; Deng, H.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B.; Chen, K. Nickel chloride (NiCl2)-caused inflammatory responses via activation of NF-κB pathway and reduction of anti-inflammatory mediator expression in the kidney. Oncotarget 2015, 6, 28607–28620. [Google Scholar] [PubMed]
- Guo, H.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B.; Chen, K.; Deng, J. Dietary NiCl2 causes G2/M cell cycle arrest in the broiler’s kidney. Oncotarget 2015, 6, 35964–35977. [Google Scholar] [PubMed]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M. Metal-induced apoptosis: Mechanisms. Mutat. Res. 2003, 533, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef] [PubMed]
- Schulze-Osthoff, K.; Ferrari, D.; Los, M.; Wesselborg, S.; Peter, M.E. Apoptosis signaling by death receptors. Eur. J. Biochem. 1998, 254, 439–459. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.M.; Oltvai, Z.N.; Yin, X.M.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993, 75, 241–251. [Google Scholar] [CrossRef]
- Chao, D.T.; Korsmeyer, S.J. Bcl-2 family: Regulators of cell death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Besbes, S.; Mirshahi, M.; Pocard, M.; Billard, C. New dimension in therapeutic targeting of Bcl-2 family proteins. Oncotarget 2015, 6, 12862–12871. [Google Scholar] [CrossRef] [PubMed]
- Martinou, J.C.; Youle, R.J. Mitochondria in apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. Bcl-2 protein family. Essential regulators of cell death. Preface. Adv. Exp. Med. Biol. 2010, 687, 7–8. [Google Scholar]
- Lavik, A.R.; Zhong, F.; Chang, M.J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388–27402. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.N.; White, M.J.; Goschnick, M.W.; Fairfax, K.A.; Tarlinton, D.M.; Kinkel, S.A.; Bouillet, P.; Adams, J.M.; Kile, B.T.; Strasser, A. Individual and overlapping roles of BH3-only proteins Bim and Bad in apoptosis of lymphocytes and platelets and in suppression of thymic lymphoma development. Cell Death Differ. 2010, 17, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Ola, M.S.; Nawaz, M.; Ahsan, H. Role of Bcl-2 family proteins and caspases in the regulation of apoptosis. Mol. Cell. Biochem. 2011, 351, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.; Green, D.R. Mechanisms of p53-dependent apoptosis. Biochem. Soc. Trans. 2001, 29, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Osaki, M.; Oshimura, M.A.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis Int. J. Program. Cell Death 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Figlin, R.A. Phosphatidylinositol-3-kinase/Akt signaling pathway and kidney cancer, and the therapeutic potential of phosphatidylinositol-3-kinase/Akt inhibitors. J. Urol. 2009, 182, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, H.; Wen, J.; Luo, K.; Liu, Q.; Huang, Y.; Zheng, Y.; Tan, Z.; Huang, Q.; Fu, J. NHE9 induces chemoradiotherapy resistance in esophageal squamous cell carcinoma by upregulating the Src/Akt/β-catenin pathway and Bcl-2 expression. Oncotarget 2015, 6, 12405–12420. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F.; Hornik, C.P.; Segev, L.; Shostak, G.A.; Sugimoto, C. PI3K/Akt and apoptosis: Size matters. Oncogene 2003, 22, 8983–8998. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.H.; Nihal, M.; Fu, V.X.; Jarrard, D.F.; Ahmad, N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3′-kinase/Akt pathway and Bcl-2 family proteins. Mol. Cancer Ther. 2006, 5, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Sun, Y.; Hu, J. Catalpol inhibits apoptosis in hydrogen peroxide-induced endothelium by activating the PI3K/Akt signaling pathway and modulating expression of Bcl-2 and Bax. Eur. J. Pharmacol. 2010, 628, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.J.; Chang, Q.S.; Wang, X.; Son, Y.O.; Liu, J.; Zhang, Z.; Bi, Y.Y.; Shi, X. Activation of Akt/GSK3β and Akt/Bcl-2 signaling pathways in nickel-transformed BEAS-2B cells. Int. J. Oncol. 2011, 39, 1285–1294. [Google Scholar] [PubMed]
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1 caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Green, D.R. Protease activation during apoptosis: Death by a thousand cuts? Cell 1995, 82, 349. [Google Scholar] [CrossRef]
- Fan, T.J.; Han, L.H.; Cong, R.S.; Liang, J. Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 2005, 37, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Bhat, M.B.; Nunez, G.; Ma, J.; Distelhorst, C.W. Regulation of Bcl-xL channel activity by calcium. J. Biol. Chem. 1998, 273, 17307–17310. [Google Scholar] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular mechanisms controlling caspase activation and function. CSH Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. CSH. Perspect. Biol. 2013, 5, a008656. [Google Scholar]
- Brentnall, M.; Rodriguez-Menocal, L.; de Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [PubMed]
- Xu, G.; Shi, Y. Apoptosis signaling pathways and lymphocyte homeostasis. Cell Res. 2007, 17, 759–771. [Google Scholar] [PubMed]
- Salvesen, G.S. Caspases: Opening the boxes and interpreting the arrows. Cell Death Differ. 2002, 9, 3–5. [Google Scholar] [CrossRef] [PubMed]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.W.; Xing, Z.; Capacio, V.L.; Peter, M.E.; Yang, X. Interdimer processing mechanism of procaspase-8 activation. EMBO J. 2003, 22, 4132–4142. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Deis, C.; Denault, J.B.; Sutherlin, D.P.; Salvesen, G.S. Activation of caspases-8 and -10 by FLIPL. Biochem. J. 2004, 382, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Stockwell, B.R.; Stennicke, H.R.; Salvesen, G.S.; Dixit, V.M. An induced proximity model for caspase-8 activation. J. Biol. Chem. 1998, 273, 2926–3290. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol. Cell 2002, 9, 459–470. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Katayama, T.; Eguchi, Y.; Kudo, T.; Taniguchi, M.; Koyama, Y.; Manabe, T.; Yamagishi, S.; Bando, Y.; Imaizumi, K.; et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. J. Cell Biol. 2004, 165, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Z.; Cao, Z.; Li, L. Allicin induces apoptosis in EL-4 cells in vitro by activation of expression of caspase-3 and -12 and up-regulation of the ratio of Bax/Bcl-2. Nat. Prod. Res. 2012, 26, 1033–1037. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, I.; Kirchhoff, S.; Krammer, P.H. Regulation of death receptor-mediated apoptosis pathways. Int. J. Biochem. 2000, 32, 1123–1136. [Google Scholar] [CrossRef]
- Locksley, R.M.; Killeen, N.; Lenardo, M.J. The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Suliman, A.; Lam, A.; Datta, R.; Srivastava, R.K. Intracellular mechanisms of TRAIL: Apoptosis through mitochondrial-dependent and -independent pathways. Oncogene 2001, 20, 2122–2133. [Google Scholar] [CrossRef] [PubMed]
- Krammer, P.H. CD95(APO-1/Fas)-mediated apoptosis: Live and let die. Adv. Immunol. 1999, 71, 163–210. [Google Scholar] [PubMed]
- Rubio-Moscardo, F.; Blesa, D.; Mestre, C.; Siebert, R.; Balasas, T.; Benito, A.; Rosenwald, A.; Climent, J.; Martinez, J.I.; Schilhabel, M.; et al. Characterization of 8p21.3 chromosomal deletions in B-cell lymphoma: TRAIL-R1 and TRAIL-R2 as candidate dosage-dependent tumor suppressor genes. Blood 2005, 106, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef] [PubMed]
- Kichev, A.; Rousset, C.I.; Baburamani, A.A.; Levison, S.W.; Wood, T.L.; Gressens, P.; Thornton, C.; Hagberg, H. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) signaling and cell death in the immature central nervous system after hypoxia-ischemia and inflammation. J. Biol. Chem. 2014, 289, 9430–9439. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.F.; Cotter, T.G. Live and let die: Regulatory mechanisms in Fas-mediated apoptosis. Cell Signal. 2003, 15, 983–992. [Google Scholar] [CrossRef]
- Dickens, L.S.; Powley, I.R.; Hughes, M.A.; MacFarlane, M. The “complexities” of life and death: Death receptor signalling platforms. Exp. Cell. Res. 2012, 318, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Lavrik, I.N.; Krammer, P.H. Regulation of CD95/Fas signaling at the DISC. Cell Death Differ. 2012, 19, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.C.; Kumar, S. Apoptosis: A mitochondrial perspective on cell death. Indian J. Exp. Biol. 2005, 43, 25–34. [Google Scholar] [PubMed]
- Logue, S.E.; Cleary, P.; Saveljeva, S.; Samali, A. New directions in ER stress-induced cell death. Apoptosis Int. J. Program. Cell Death 2013, 18, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Vaux, D.L. Apoptogenic factors released from mitochondria. Biochim. Biophys. Acta 2011, 1813, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar] [CrossRef]
- Duckett, C.S. IAP proteins: Sticking it to Smac. Biochem. J. 2005, 385, e1–e2. [Google Scholar] [CrossRef] [PubMed]
- Garrido, C.; Galluzzi, L.; Brunet, M.; Puig, P.E.; Didelot, C.; Kroemer, G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006, 13, 1423–1433. [Google Scholar] [CrossRef] [PubMed]
- Vande Walle, L.; Lamkanfi, M.; Vandenabeele, P. The mitochondrial serine protease HtrA2/Omi: An overview. Cell Death Differ. 2008, 15, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Reed, J.C. IAP family proteins—Suppressors of apoptosis. Genes Dev. 1999, 13, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Suzuki, K.; Sakamoto, C.; Ohta, K.; Nishiki, S.; Hino, M.; Tatsumi, N.; Kitagawa, S. Expression of the inhibitor of apoptosis (IAP) family members in human neutrophils: Up-regulation of cIAP2 by granulocyte colony-stimulating factor and overexpression of cIAP2 in chronic neutrophilic leukemia. Blood 2003, 101, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Chai, J.; Suber, T.L.; Wu, J.W.; Du, C.; Wang, X.; Shi, Y. Structural basis of IAP recognition by Smac/DIABLO. Nature 2000, 408, 1008–1012. [Google Scholar] [PubMed]
- Norberg, E.; Orrenius, S.; Zhivotovsky, B. Mitochondrial regulation of cell death: Processing of apoptosis-inducing factor (AIF). Biochem. Biophys Res. Commun. 2010, 396, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Buttner, S.; Eisenberg, T.; Carmona-Gutierrez, D.; Ruli, D.; Knauer, H.; Ruckenstuhl, C.; Sigrist, C.; Wissing, S.; Kollroser, M.; Frohlich, K.U.; et al. Endonuclease G regulates budding yeast life and death. Mol. Cell 2007, 25, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Van Loo, G.; Schotte, P.; van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Urra, H.; Dufey, E.; Lisbona, F.; Rojas-Rivera, D.; Hetz, C. When ER stress reaches a dead end. Biochim. Biophys. Acta 2013, 1833, 3507–3517. [Google Scholar] [CrossRef] [PubMed]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H. ER stress and diseases. FEBS J. 2007, 274, 630–658. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, N.; Sugiyama, Y.; Miyazaki, S.; Nakagawa, H.; Nishimura, K.; Matsuo, S. An ATF4-signal-modulating machine other than GADD34 acts in ATF4-to-CHOP signaling to block CHOP expression in ER-stress-related autophagy. J. Cell. Biochem. 2015, 116, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Yoshii, S.; Hattori, T.; Onozaki, K.; Hayashi, H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005, 24, 1243–1255. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.D.; Bultynck, G.; Rizzuto, R.; Decuypere, J.P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.A. ER stress signaling and the Bcl-2 family of proteins: From adaptation to irreversible cellular damage. Antioxid. Redox Signal. 2007, 9, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.L.; Wu, J.L.; Chen, M.H.; Hong, J.R. Aquatic birnavirus-induced ER stress-mediated death signaling contribute to downregulation of Bcl-2 family proteins in salmon embryo cells. PLoS ONE 2011, 6, e22935. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Yang, W.; He, L.J.; Ding, W.J.; Zheng, L.; Liao, S.D.; Huang, P.; Lu, W.; He, Q.J.; Yang, B. Upregulating Noxa by ER stress, celastrol exerts synergistic anti-cancer activity in combination with ABT-737 in human hepatocellular carcinoma cells. PLoS ONE 2012, 7, e52333. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Kirschnek, S.; Wiesmeier, M.; Vier, J.; Hacker, G. Roscovitine-induced apoptosis in neutrophils and neutrophil progenitors is regulated by the Bcl-2-family members Bim, Puma, Noxa and Mcl-1. PLoS ONE 2013, 8, e79352. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Jurczak, M.J.; Lee, A.H.; Jornayvaz, F.R.; Lee, H.Y.; Birkenfeld, A.L.; Guigni, B.A.; Kahn, M.; Samuel, V.T.; Glimcher, L.H.; Shulman, G.I. Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice. J. Biol. Chem. 2012, 287, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Furuhata, M.; Takada, E.; Noguchi, T.; Ichijo, H.; Mizuguchi, J. Apoptosis signal-regulating kinase (ASK)-1 mediates apoptosis through activation of JNK1 following engagement of membrane immunoglobulin. Exp. Cell Res. 2009, 315, 3467–3476. [Google Scholar] [CrossRef] [PubMed]
- Newton, V.L.; Ali, S.; Duddy, G.; Whitmarsh, A.J.; Gardiner, N.J. Targeting apoptosis signalling kinase-1 (ASK-1) does not prevent the development of neuropathy in streptozotocin-induced diabetic mice. PLoS ONE 2014, 9, e107437. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Robert Parker, J.M.; Opas, M. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium 2002, 32, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Timmins, J.M.; Ozcan, L.; Seimon, T.A.; Li, G.; Malagelada, C.; Backs, J.; Backs, T.; Bassel-Duby, R.; Olson, E.N.; Anderson, M.E.; et al. Calcium/calmodulin-dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J. Clin. Investig. 2009, 119, 2925–2941. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2143–2152. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Yuan, J. Cross-talk between two cysteine protease families: Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000, 150, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Koenig, U.; Eckhart, L.; Tschachler, E. Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun. 2002, 293, 722–726. [Google Scholar] [CrossRef]
- Bian, Z.M.; Elner, S.G.; Elner, V.M. Dual involvement of caspase-4 in inflammatory and ER stress-induced apoptotic responses in human retinal pigment epithelial cells. Investig. Ophthalmol. Vis. Sci. 2009, 50, 6006–6014. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Deng, Y.; Zhang, Y.; Li, C.; Zhang, R.; Sun, Y.; Zhang, K.; Li, J.; Yao, S. Protective effects of grape seed procyanidin extract against nickel sulfate-induced apoptosis and oxidative stress in rat testes. Toxicol. Mech. Methods 2011, 21, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.M.; Zheng, G.H.; Ming, Q.L.; Chao, C.; Sun, J.M. Sesamin protects mouse liver against nickel-induced oxidative DNA damage and apoptosis by the PI3K-Akt pathway. J. Agric. Food Chem. 2013, 61, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.H.; Liu, C.M.; Sun, J.M.; Feng, Z.J.; Cheng, C. Nickel-induced oxidative stress and apoptosis in Carassius auratus liver by JNK pathway. Aquat. Toxicol. 2014, 147, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem. Toxicol. 2014, 63, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wu, B. The association between splenocyte apoptosis and alterations of Bax, Bcl-2 and caspase-3 mRNA expression, and oxidative stress induced by dietary nickel chloride in broilers. Int. J. Environ. Res. Public Health 2013, 10, 7310–7326. [Google Scholar] [CrossRef] [PubMed]
- Nowak, M.; Kopp, F.; Roelofs-Haarhuis, K.; Wu, X.; Gleichmann, E. Oral nickel tolerance: Fas ligand-expressing invariant NK T cells promote tolerance induction by eliciting apoptotic death of antigen-carrying, effete B cells. J. Immunol. 2006, 176, 4581–4589. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Zhang, D.; Wang, X.; Chen, J. Nitric oxide and Bcl-2 mediated the apoptosis induced by nickel(II) in human T hybridoma cells. Toxicol. Appl. Pharmacol. 2007, 221, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Zhang, D.; Chen, J.; Lin, C.; Liu, Q. Involvement of histone hypoacetylation in Ni2+-induced Bcl-2 down-regulation and human hepatoma cell apoptosis. J. Biol. Inorg. Chem. 2004, 9, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Cavani, A. Breaking tolerance to nickel. Toxicology 2005, 209, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Ahamed, M.; Ahmad, J.; Majeed Khan, M.A.; Musarrat, J.; Al-Khedhairy, A.A.; Alrokayan, S.A. Nickel oxide nanoparticles induce cytotoxicity, oxidative stress and apoptosis in cultured human cells that is abrogated by the dietary antioxidant curcumin. Food Chem. Toxicol. 2012, 50, 641–647. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Ali, D.; Alhadlaq, H.A.; Akhtar, M.J. Nickel oxide nanoparticles exert cytotoxicity via oxidative stress and induce apoptotic response in human liver cells (HepG2). Chemosphere 2013, 93, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.; Barcellos-de-Souza, P.; Barja-Fidalgo, C.; Fernandes, E. Nickel induces apoptosis in human neutrophils. Biometals Int. J. Role Metal Ions Biol. Biochem. Med. 2013, 26, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Wang, Y.F.; Huang, W.R.; Huang, Y.T. Nickel induces oxidative stress and genotoxicity in human lymphocytes. Toxicol. Appl. Pharmacol. 2003, 189, 153–159. [Google Scholar] [CrossRef]
- Shiao, Y.H.; Lee, S.H.; Kasprzak, K.S. Cell cycle arrest, apoptosis and p53 expression in nickel(II) acetate-treated Chinese hamster ovary cells. Carcinogenesis 1998, 19, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Dallas, L.J.; Bean, T.P.; Turner, A.; Lyons, B.P.; Jha, A.N. Oxidative DNA damage may not mediate Ni-induced genotoxicity in marine mussels: Assessment of genotoxic biomarkers and transcriptional responses of key stress genes. Mutat. Res. 2013, 754, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef] [PubMed]
- Avery, S.V. Molecular targets of oxidative stress. Biochem. J. 2011, 434, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis Int. J. Program. Cell Death 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Morris, H.; Cronin, M. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Song, M.; Zhang, Y.; Yan, M.; Zhang, M.; Bi, H. Nickel nanowires induce cell cycle arrest and apoptosis by generation of reactive oxygen species in HeLa cells. Toxicol. Rep. 2014, 1, 114–121. [Google Scholar] [CrossRef]
- Pan, J.; Chang, Q.; Wang, X.; Son, Y.; Zhang, Z.; Chen, G.; Luo, J.; Bi, Y.; Chen, F.; Shi, X. Reactive oxygen species-activated Akt/ASK1/p38 signaling pathway in nickel compound-induced apoptosis in BEAS 2B cells. Chem. Res. Toxicol. 2010, 23, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M. Toxic response of nickel nanoparticles in human lung epithelial A549 cells. Toxicol. Vitro 2011, 25, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Kubrak, O.I.; Husak, V.V.; Rovenko, B.M.; Poigner, H.; Mazepa, M.A.; Kriews, M.; Abele, D.; Lushchak, V.I. Tissue specificity in nickel uptake and induction of oxidative stress in kidney and spleen of goldfish Carassius auratus, exposed to waterborne nickel. Aquat. Toxicol. 2012, 118–119, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride induces oxidative intestinal damage in broilers. Int. J. Environ. Res. Publ. Health 2013, 10, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, G. Oxidative damage effects in the copepod Tigriopus japonicus Mori experimentally exposed to nickel. Ecotoxicology 2010, 19, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Attig, H.; Kamel, N.; Sforzini, S.; Dagnino, A.; Jamel, J.; Boussetta, H.; Viarengo, A.; Banni, M. Effects of thermal stress and nickel exposure on biomarkers responses in Mytilus galloprovincialis (Lam). Mar. Environ. Res. 2014, 94, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Alarifi, S.; Ali, D.; Alakhtani, S.; Al Suhaibani, E.S.; Al-Qahtani, A.A. Reactive oxygen species-mediated DNA damage and apoptosis in human skin epidermal cells after exposure to nickel nanoparticles. Biol. Trace Elem. Res. 2014, 157, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.B.; Messer, R.L.; McCloud, V.V.; Lockwood, P.E.; Hsu, S.D.; Wataha, J.C. Ni(II) activates the Nrf2 signaling pathway in human monocytic cells. Biomaterials 2006, 27, 5348–5356. [Google Scholar] [CrossRef] [PubMed]
- Krezel, A.; Szczepanik, W.; Sokolowska, M.; Jezowska-Bojczuk, M.; Bal, W. Correlations between complexation modes and redox activities of Ni(II)-GSH complexes. Chem. Res. Toxicol. 2003, 16, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bowman, L.; Zhang, X.; Shi, X.; Jiang, B.; Castranova, V.; Ding, M. Metallic nickel nano- and fine particles induce JB6 cell apoptosis through a caspase-8/AIF mediated cytochrome c-independent pathway. J. Nanobiotechnol. 2009, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Bonin, S.; Larese, F.F.; Trevisan, G.; Avian, A.; Rui, F.; Stanta, G.; Bovenzi, M. Gene expression changes in peripheral blood mononuclear cells in occupational exposure to nickel. Exp. Dermatol. 2011, 20, 147–148. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Zamzami, N.; Castedo, M.; Hirsch, T.; Marchetti, P.; Macho, A.; Daugas, E.; Geuskens, M.; Kroemer, G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996, 184, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Shyu, H.W.; Chang, Y.C.; Tseng, W.C.; Huang, Y.L.; Lin, K.H.; Chou, M.C.; Liu, H.L.; Chen, C.Y. Nickel (II)-induced cytotoxicity and apoptosis in human proximal tubule cells through a ROS- and mitochondria-mediated pathway. Toxicol. Appl. Pharmacol. 2012, 259, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Ahamed, M.; Akhtar, M.J.; Alhadlaq, H.A.; Khan, M.A.; Alrokayan, S.A. Comparative cytotoxic response of nickel ferrite nanoparticles in human liver HepG2 and breast MFC-7 cancer cells. Chemosphere 2015, 135, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Patel, E.; Lynch, C.; Ruff, V.; Reynolds, M. Co-exposure to nickel and cobalt chloride enhances cytotoxicity and oxidative stress in human lung epithelial cells. Toxicol. Appl. Pharmacol. 2012, 258, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Duan, W.X.; He, M.D.; Mao, L.; Qian, F.H.; Li, Y.M.; Pi, H.F.; Liu, C.; Chen, C.H.; Lu, Y.H.; Cao, Z.W.; et al. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol. Appl. Pharmacol. 2015, 286, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Buschini, A.; Pinelli, S.; Pellacani, C.; Giordani, F.; Ferrari, M.B.; Bisceglie, F.; Giannetto, M.; Pelosi, G.; Tarasconi, P. Synthesis, characterization and deepening in the comprehension of the biological action mechanisms of a new nickel complex with antiproliferative activity. J. Inorg. Biochem. 2009, 103, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Hiramatsu, N.; Kasai, A.; Du, S.; Takeda, M.; Hayakawa, K.; Okamura, M.; Yao, J.; Kitamura, M. Rapid, transient induction of ER stress in the liver and kidney after acute exposure to heavy metal: Evidence from transgenic sensor mice. FEBS Lett. 2007, 581, 2055–2059. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, B.; Liebermann, D.A. Apoptotic signaling by c-Myc. Oncogene 2008, 27, 6462–6472. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.A.; Cleveland, J.L. Myc pathways provoking cell suicide and cancer. Oncogene 2003, 22, 9007–9021. [Google Scholar] [CrossRef] [PubMed]
- Juin, P.; Hueber, A.O.; Littlewood, T.; Evan, G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999, 13, 1367–1381. [Google Scholar] [CrossRef] [PubMed]
- Eischen, C.M.; Woo, D.; Roussel, M.F.; Cleveland, J.L. Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 2001, 21, 5063–5070. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Suen, T.C.; Sun, H.; Arita, A.; Costa, M. Nickel compounds induce apoptosis in human bronchial epithelial Beas-2B cells by activation of c-Myc through ERK pathway. Toxicol. Appl. Pharmacol. 2009, 235, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting Bcl2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Hengartner, M.O. miRNAs and apoptosis: RNAs to die for. Oncogene 2006, 25, 6176–6187. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Busacca, S.; Almeida, G.M.; Gaudino, G.; Fennell, D.A.; Vasconcelos, M.H. MicroRNA regulation of core apoptosis pathways in cancer. Eur. J. Cancer 2011, 47, 163–174. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, H.; Chen, L.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wang, X.; Wu, B. Research Advances on Pathways of Nickel-Induced Apoptosis. Int. J. Mol. Sci. 2016, 17, 10. https://doi.org/10.3390/ijms17010010
Guo H, Chen L, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Wu B. Research Advances on Pathways of Nickel-Induced Apoptosis. International Journal of Molecular Sciences. 2016; 17(1):10. https://doi.org/10.3390/ijms17010010
Chicago/Turabian StyleGuo, Hongrui, Lian Chen, Hengmin Cui, Xi Peng, Jing Fang, Zhicai Zuo, Junliang Deng, Xun Wang, and Bangyuan Wu. 2016. "Research Advances on Pathways of Nickel-Induced Apoptosis" International Journal of Molecular Sciences 17, no. 1: 10. https://doi.org/10.3390/ijms17010010
APA StyleGuo, H., Chen, L., Cui, H., Peng, X., Fang, J., Zuo, Z., Deng, J., Wang, X., & Wu, B. (2016). Research Advances on Pathways of Nickel-Induced Apoptosis. International Journal of Molecular Sciences, 17(1), 10. https://doi.org/10.3390/ijms17010010