
APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome

 ,
,  ,
, 

Abstract
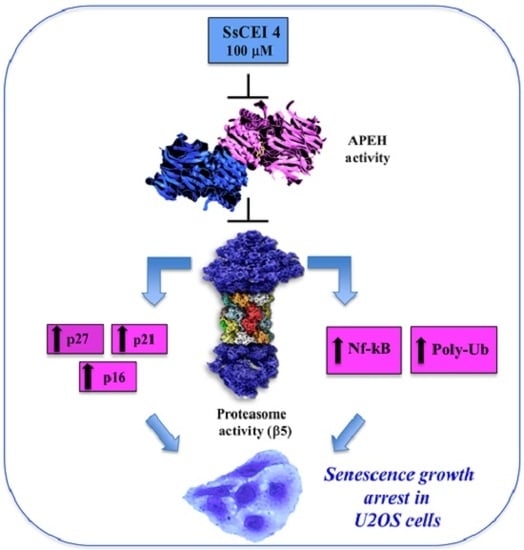
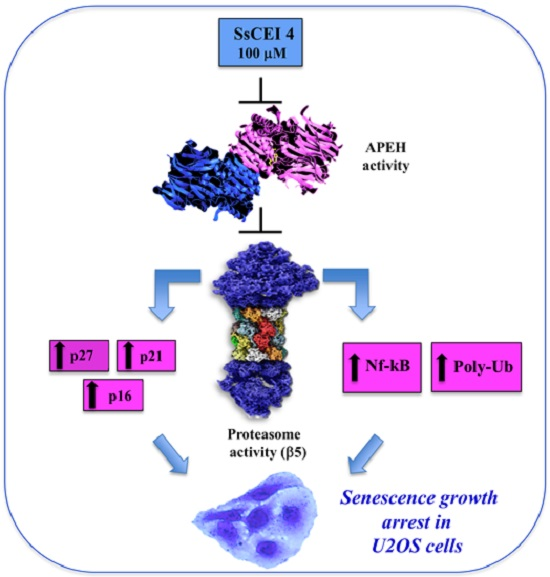
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. SsCEI 4 Is Able to Inhibit Acylpeptide Hydrolase (APEH) Activity but Not Proteasome in Cell-Free Assays
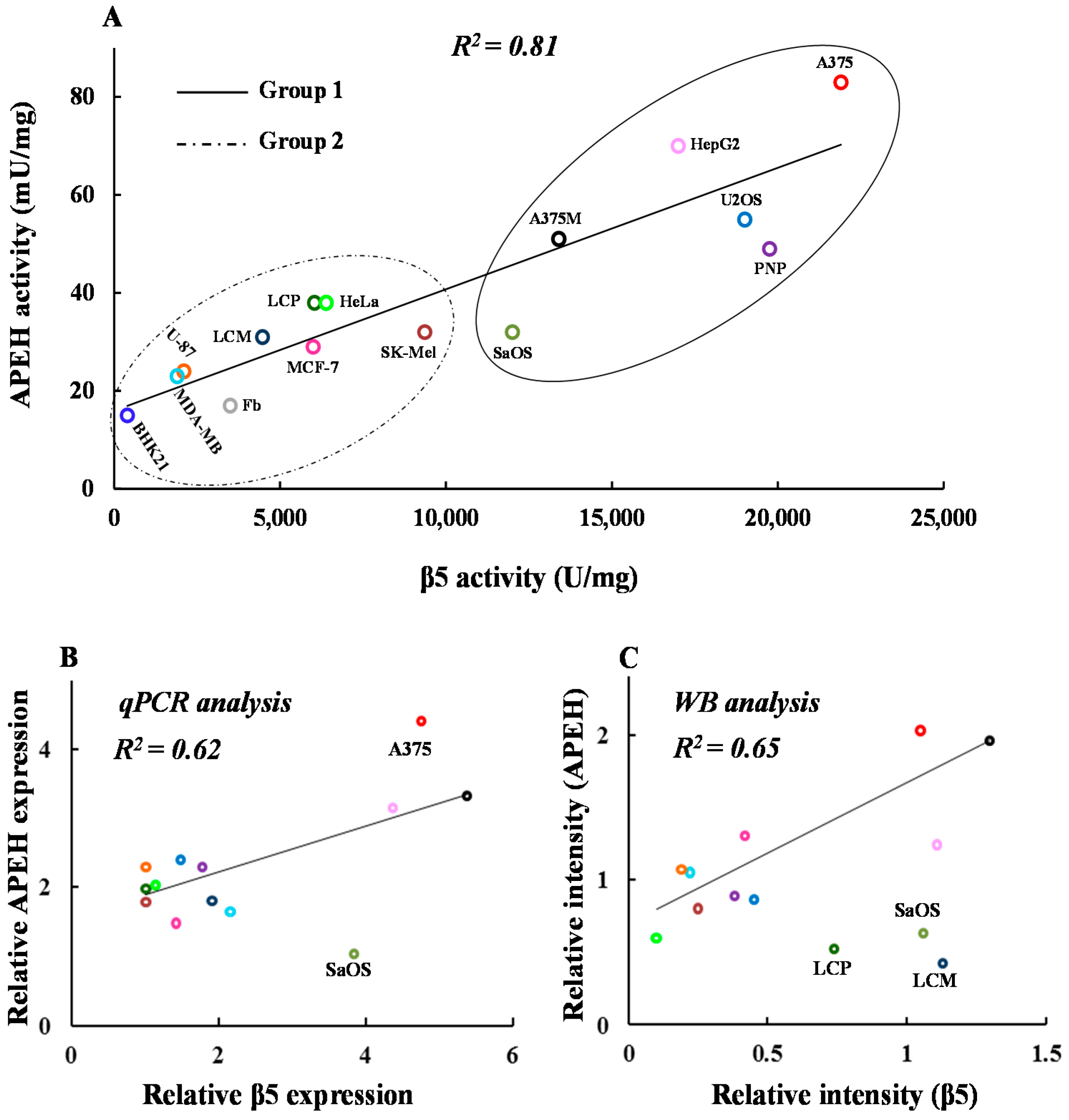
2.2. mRNA, Activity and Protein Levels of APEH, and Proteasome Correlate in a Wide Panel of Cancer Cell Lines
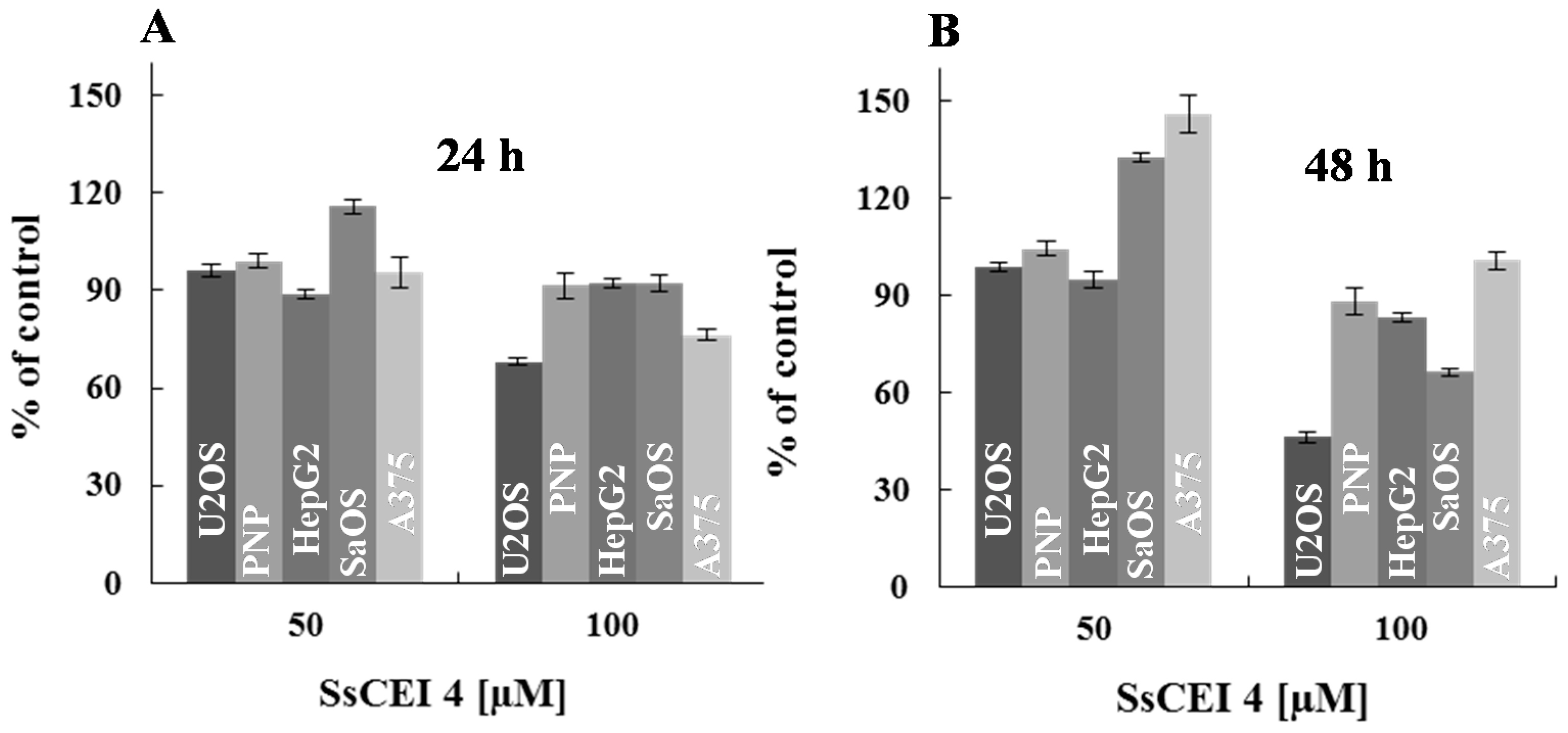
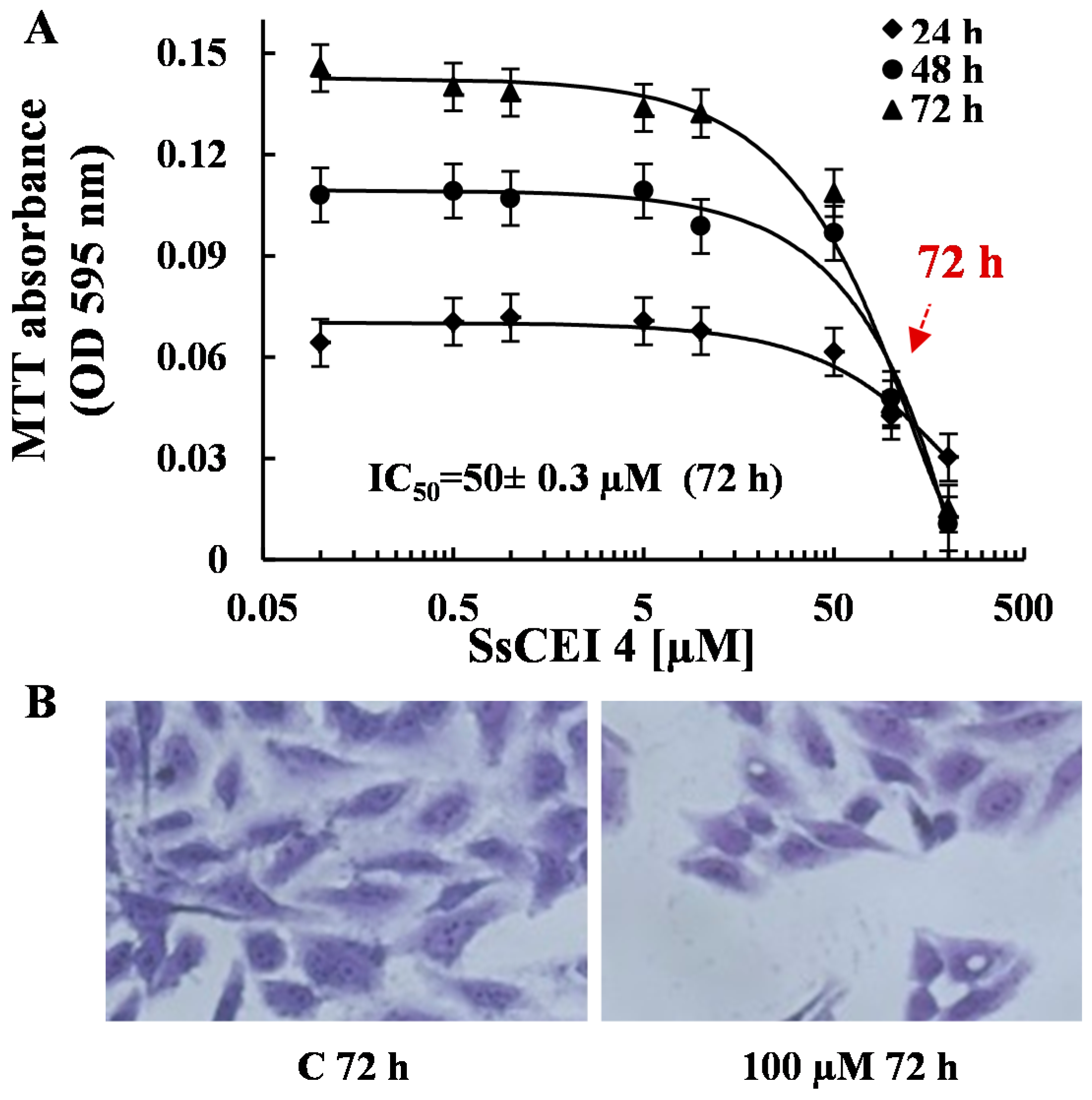
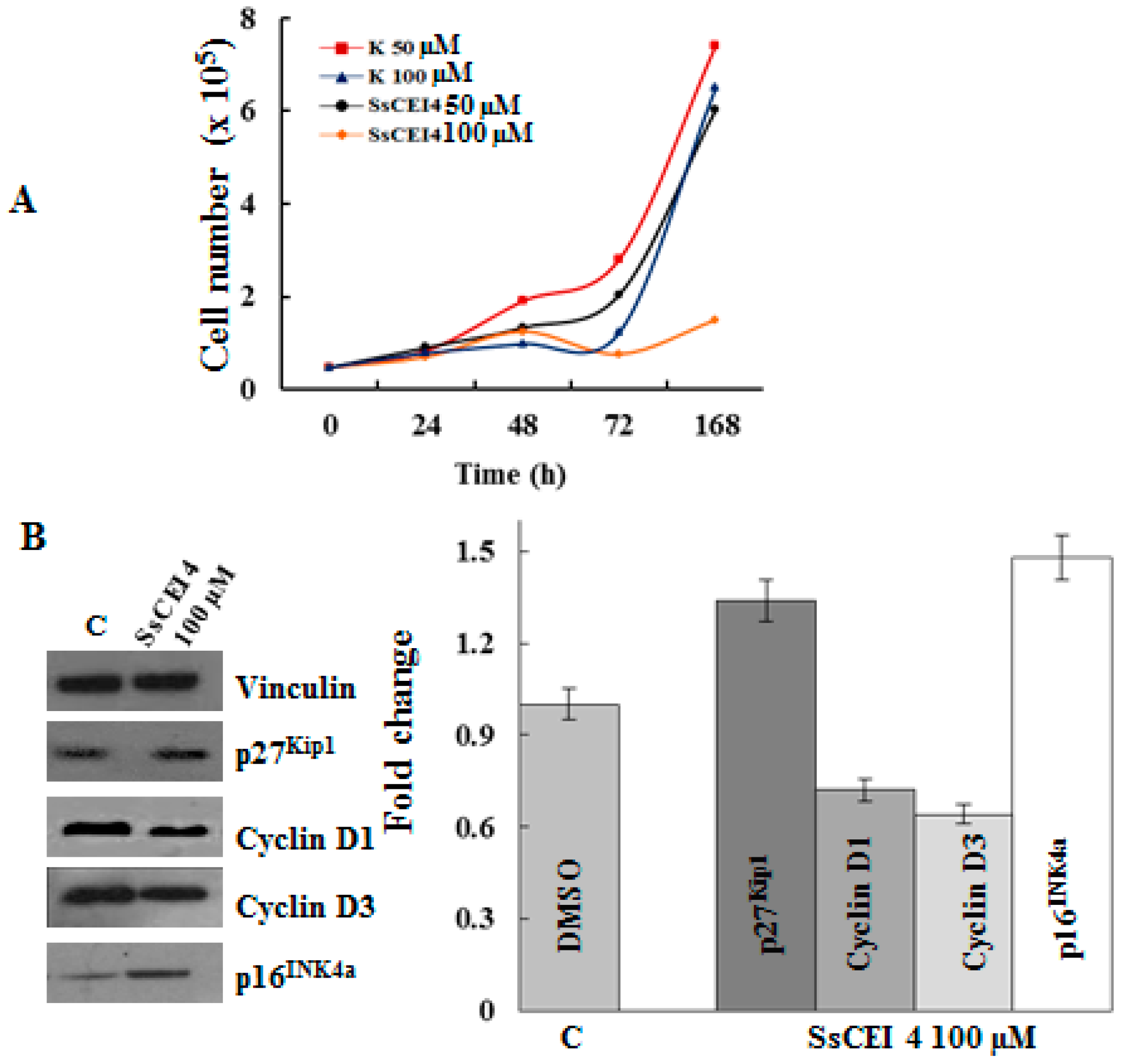
2.3. SsCEI 4 Is Effective in Reducing Osteosarcoma Cell Viability in a Dose- and Time-Dependent Manner
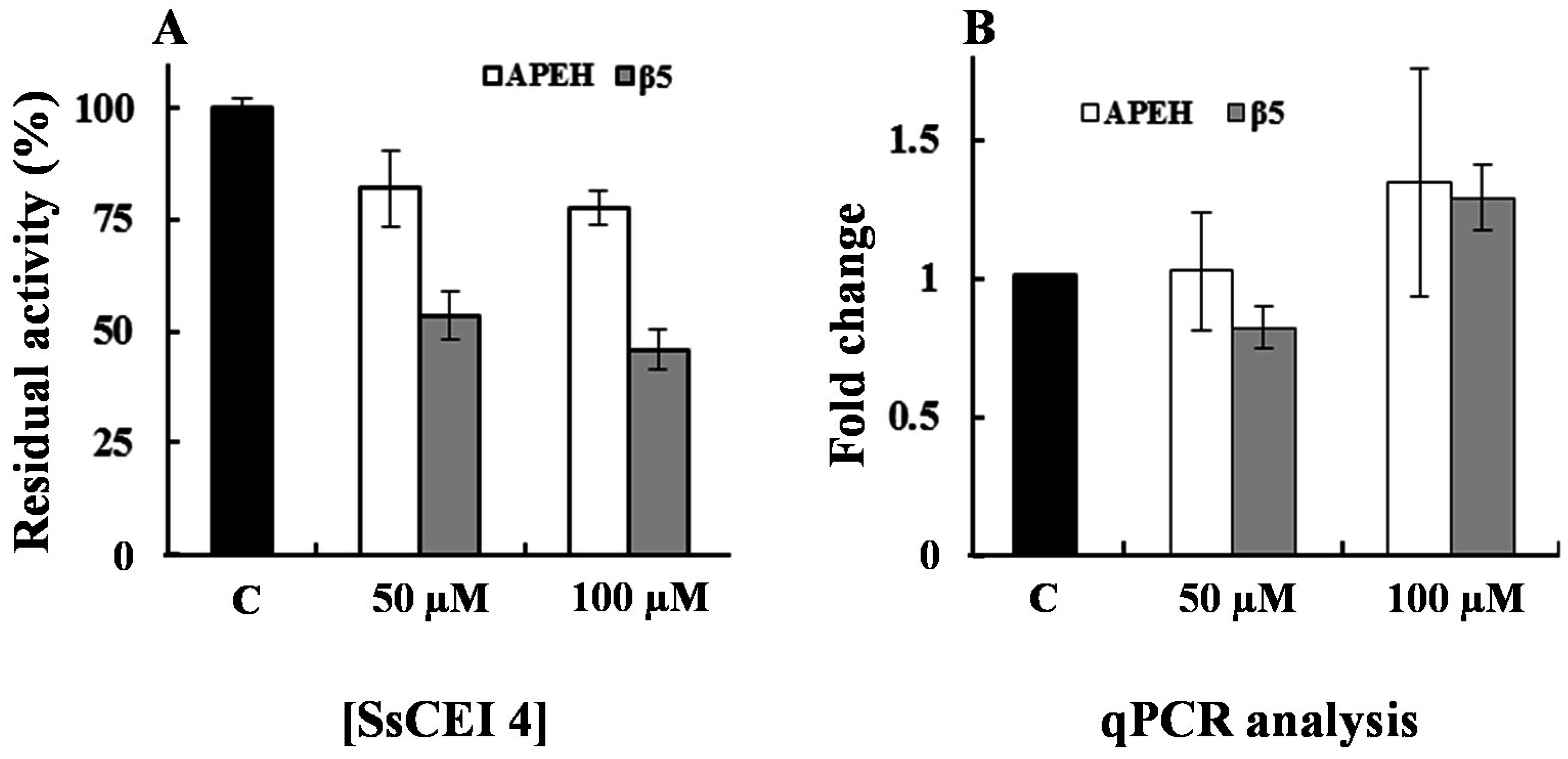
2.4. SsCEI 4 Induces APEH-Proteasome Downregulation in U2OS Cells
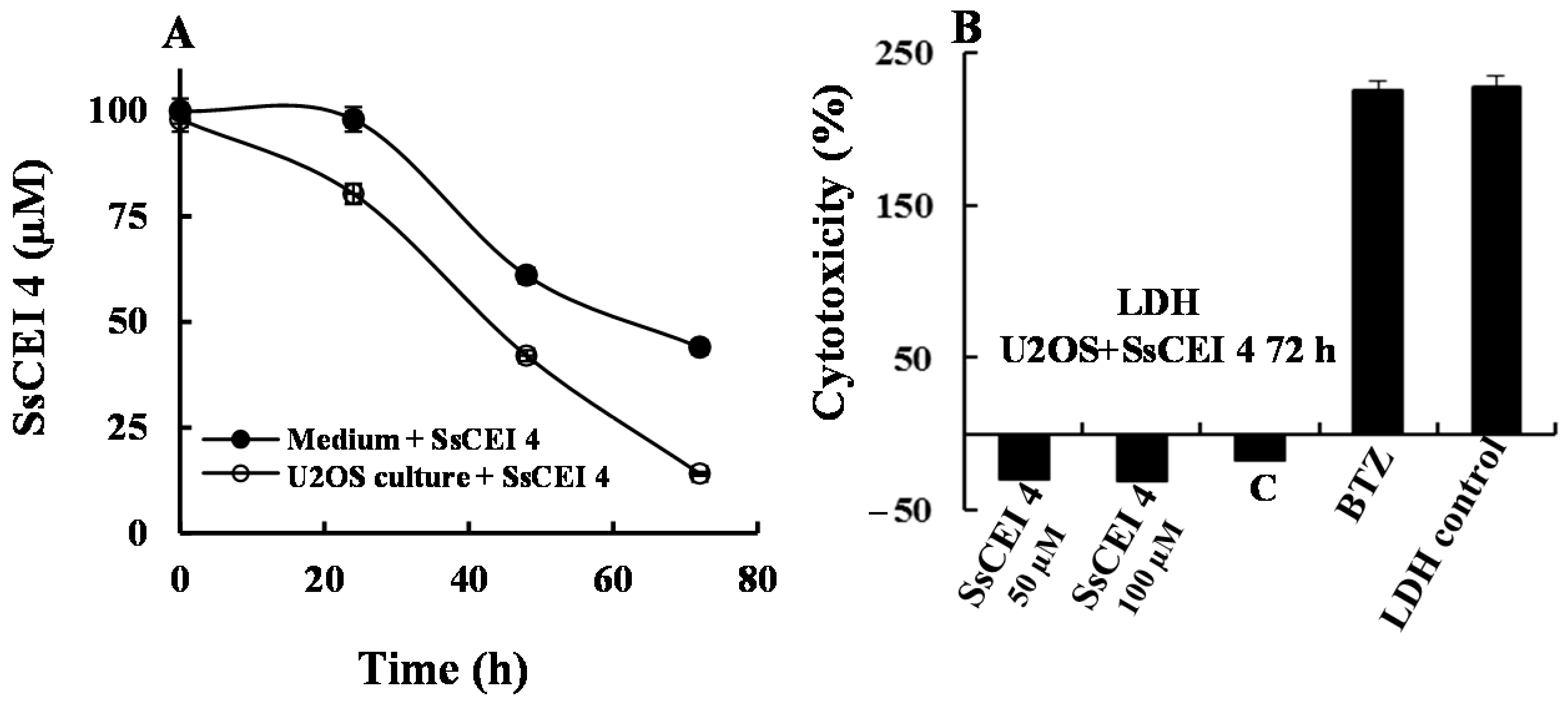
2.5. SsCEI 4 Treatment of U2OS Does Not Induce Cytotoxicity, Apoptosis, or Autophagy
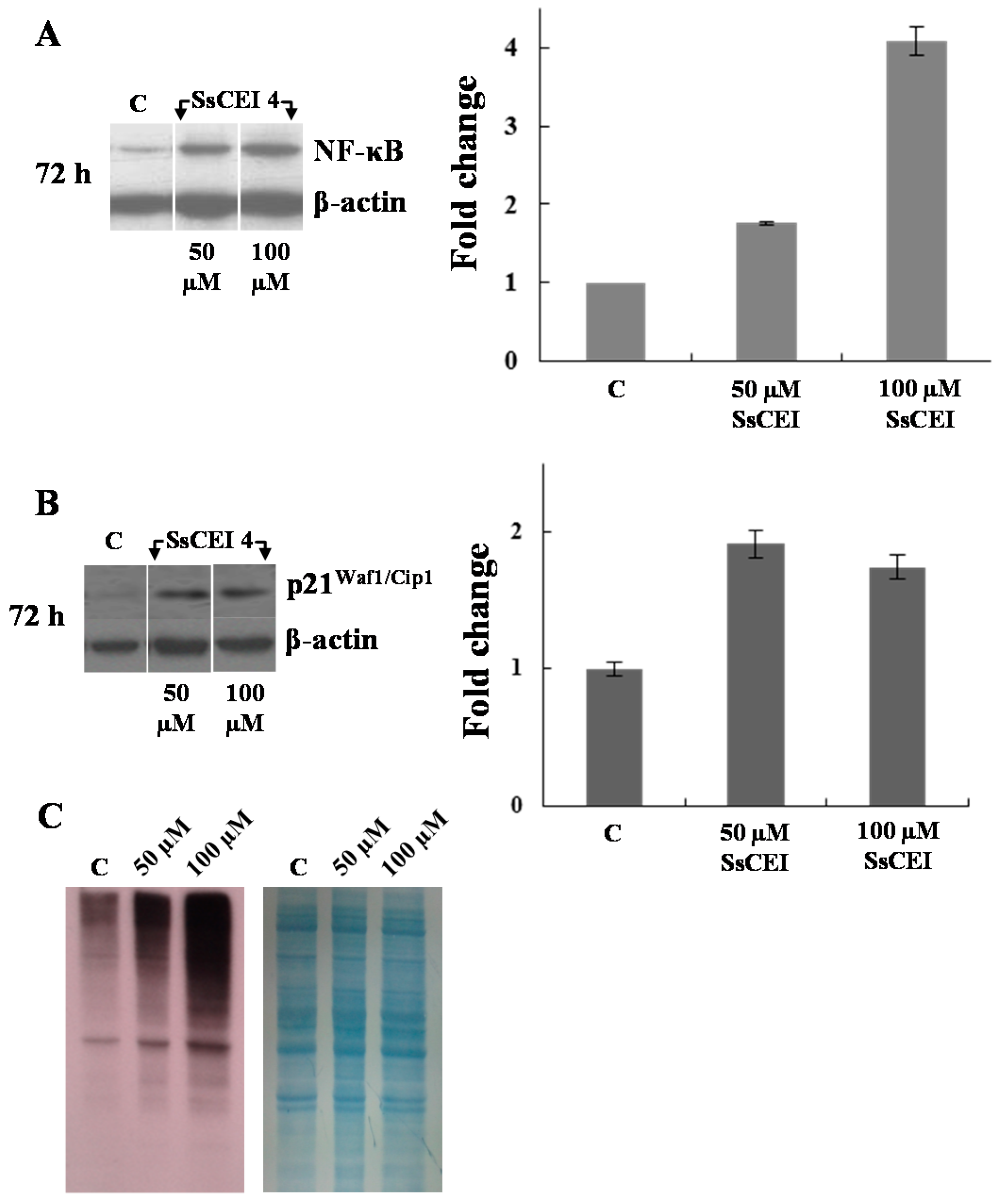
2.6. SsCEI 4 Treatment Promotes Senescence and Alteration of Cell Cycle Regulators in U2OS Cells
3. Materials and Methods
3.1. Reagents
3.2. Enzyme Assays
3.3. Cells, Culture Conditions and Treatments
3.4. Protein Extraction and Western Blotting Analyses
3.5. MTT Assay and Crystal Violet Staining
3.6. Apoptosis Assays
3.7. Cytotoxicity Assay
3.8. Cellular Internalization Assay of SsCEI 4 Peptide
3.9. Quantitative Real-Time PCR (qPCR) Analysis
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Field-Smith, A.; Morgan, G.J.; Davies, F.E. Bortezomib (Velcade™) in the Treatment of Multiple Myeloma. Ther. Clin. Risk Manag. 2006, 2, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Merin, N.M.; Kelly, K.R. Clinical use of proteasome inhibitors in the treatment of multiple myeloma. Pharmaceuticals (Basel) 2014, 8, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Richardson, P.G.; Cavo, M.; Orlowski, R.Z.; San Miguel, J.F.; Palumbo, A.; Harousseau, J.L. Proteasome inhibitors in multiple myeloma: 10 years later. Blood 2012, 120, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.K.; Cho, C.H.; Lee, C.W.; Wu, K.; Fan, D.; Yu, J.; Sung, J.J. Proteasome inhibition: A new therapeutic strategy to cancer treatment. Cancer Lett. 2010, 293, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, R.Z.; Kuhn, D.J. Proteasome inhibitors in cancer therapy: Lessons from the first decade. Clin. Cancer Res. 2008, 14, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Matondo, M.; Bousquet-Dubouch, M.P.; Gallay, N.; Uttenweiler-Joseph, S.; Recher, C.; Payrastre, B.; Manenti, S.; Monsarrat, B.; Burlet-Schiltz, O. Proteasome inhibitor-induced apoptosis in acute myeloid leukemia: A correlation with the proteasome status. Leuk. Res. 2010, 34, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Dou, Q.P. The ubiquitin-proteasome system as a prospective molecular target for cancer treatment and prevention. Curr. Protein Pept. Sci. 2010, 11, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y. Structure, assembly and homeostatic regulation of the 26S proteasome. J. Mol. Cell. Biol. 2010, 2, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Taylor, A. Ubiquitin-proteasome pathway and cellular responses to oxidative stress. Free Radic. Biol. Med. 2011, 51, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L. Protein degradation and protection against misfolded or damaged proteins. Nature 2003, 426, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Kisselev, A.F.; Goldberg, A.L. Proteasome inhibitors: From research tools to drug candidates. Chem. Biol. 2001, 8, 739–758. [Google Scholar] [CrossRef]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Miller, C.P.; Ban, K.; Dujka, M.E.; McConkey, D.J.; Munsell, M.; Palladino, M.; Chandra, J. NPI-0052, a novel proteasome inhibitor, induces caspase-8 and ROS-dependent apoptosis alone and in combination with HDAC inhibitors in leukemia cells. Blood 2007, 110, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.E.; Davenport, E.L.; Smith, E.M.; Muralikrishnan, S.; Dunlop, A.S.; Walker, B.A.; Krige, D.; Drummond, A.H.; Hooftman, L.; Morgan, G.J.; et al. Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol. Cancer Ther. 2009, 8, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, P.; Cocca, E.; Palumbo, R.; Gogliettino, M.; Rossi, M.; Palmieri, G. RedOx status, proteasome and APEH: Insights into anticancer mechanisms of t10,c12-conjugated linoleic acid isomer on A375 melanoma cells. PLoS ONE 2013, 8, e80900. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, G.; Bergamo, P.; Luini, A.; Ruvo, M.; Gogliettino, M.; Langella, E.; Saviano, M.; Hegde, R.N.; Sandomenico, A.; Rossi, M. Acylpeptide hydrolase inhibition as targeted strategy to induce proteasomal down-regulation. PLoS ONE 2011, 6, e25888. [Google Scholar] [CrossRef] [PubMed]
- Polgár, L. The prolyl oligopeptidase family. Cell. Mol. Life Sci. 2002, 59, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.; Ortwerth, B.J. Bovine lens acylpeptide hydrolase. Purification and characterization of a tetrameric enzyme resistant to urea denaturation and proteolytic inactivation. Eur. J. Biochem. 1993, 216, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Brunialti, E.A.; Gatti-Lafranconi, P.; Lotti, M. Promiscuity, stability and cold adaptation of a newly isolated acylaminoacyl peptidase. Biochimie 2011, 93, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Gogliettino, M.; Balestrieri, M.; Cocca, E.; Mucerino, S.; Rossi, M.; Petrillo, M.; Mazzella, E.; Palmieri, G. Identification and characterisation of a novel acylpeptide hydrolase from Sulfolobus solfataricus: Structural and functional insights. PLoS ONE 2012, 7, e37921. [Google Scholar] [CrossRef] [PubMed]
- Nakai, A.; Yamauchi, Y.; Sumi, S.; Tanaka, K. Role of acylamino acid-releasing enzyme/oxidized protein hydrolase in sustaining homeostasis of the cytoplasmic antioxidative system. Planta 2012, 236, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Gogliettino, M.; Riccio, A.; Balestrieri, M.; Cocca, E.; Facchiano, A.; D’Arco, T.M.; Tesoro, C.; Rossi, M.; Palmieri, G. A novel class of bifunctional acylpeptide hydrolases—Potential role in the antioxidant defense systems of the Antarctic fish Trematomus bernacchii. FEBS J. 2014, 281, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Riccio, A.; Gogliettino, M.; Palmieri, G.; Balestrieri, M.; Facchiano, A.; Rossi, M.; Palumbo, S.; Monti, G.; Cocca, E. A New APEH Cluster with Antioxidant Functions in the Antarctic Hemoglobinless Icefish Chionodraco hamatus. PLoS ONE 2015, 10, e0125594. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kiuchi, Y.; Ando, K.; Hayakawa, M.; Kikugawa, K. Coordination of oxidized protein hydrolase and the proteasome in the clearance of cytotoxic denatured proteins. Biochem. Biophys. Res. Commun. 2004, 324, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Watanabe, K.; Beppu, M.; Kikugawa, K.; Yasuda, H. Identification of oxidized protein hydrolase of human erythrocytes as acylpeptide hydrolase. Biochim. Biophys. Acta 2000, 1478, 102–112. [Google Scholar] [CrossRef]
- Fujino, T.; Tada, T.; Beppu, M.; Kikugawa, K. Purification and characterization of a serine protease in erythrocyte cytosol that is adherent to oxidized membranes and preferentially degrades proteins modified by oxidation and glycation. J. Biochem. 1998, 124, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- López-Granero, C.; Cardona, D.; Giménez, E.; Lozano, R.; Barril, J.; Aschner, M.; Sánchez-Santed, F.; Cañadas, F. Comparative study on short- and long-term behavioral consequences of organophosphate exposure: Relationship to AChE mRNA expression. Neurotoxicology 2014, 40, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Zhao, J.; Collins, G.A. Blocking Cancer Growth with Less POMP or Proteasomes. Mol. Cell 2015, 59, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Almond, J.B.; Cohen, G.M. The proteasome: A novel target for cancer chemotherapy. Leukemia 2002, 16, 433–443. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a026716. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N.O.; Tafani, M.; Schneider, T.; Russo, M.A.; Farber, J.L. The course of etoposide-induced apoptosis in Jurkat cells lacking p53 and Bax. J. Cell. Physiol. 2006, 208, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, I.; Russo, M.; Bilotto, S.; Spagnuolo, C.; Scognamiglio, A.; Palumbo, R.; Nappo, A.; Iacomino, G.; Moio, L.; Russo, G.L. Dealcoholated red wine induces autophagic and apoptotic cell death in an osteosarcoma cell line. Food Chem. Toxicol. 2013, 60, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Adams, J. The Proteasome in Cell-Cycle Regulation. In Proteasome Inhibitors in Cancer Therapy; Adams, J., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 77–84. [Google Scholar]
- Dai, C.Y.; Enders, G.H. p16INK4a can initiate an autonomous senescence program. Oncogene 2000, 19, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tong, T. FOXA1 antagonizes EZH2-mediated CDKN2A repression in carcinogenesis. Biochem. Biophys. Res. Commun. 2014, 453, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- King, R.W.; Deshaies, R.J.; Peters, J.M.; Kirschner, M.W. How proteolysis drives the cell cycle. Science 1996, 274, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.I. The ubiquitin-proteasome pathway in cell cycle control. Results Probl. Cell Differ. 2006, 42, 147–181. [Google Scholar] [PubMed]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system-implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Rivett, A.J.; Gardner, R.C. Proteasome inhibitors: From in vitro uses to clinical trials. J. Pept. Sci. 2000, 6, 478–488. [Google Scholar] [CrossRef]
- Althubiti, M.; Lezina, L.; Carrera, S.; Jukes-Jones, R.; Giblett, S.M.; Antonov, A.; Barlev, N.; Saldanha, G.S.; Pritchard, C.A.; Cain, K.; et al. Characterization of novel markers of senescence and their prognostic potential in cancer. Cell Death Dis. 2014, 5, e1528. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Universal ProbeLibrary Assay Design Center (Roche Diagnostics). Available online: https://lifescience.roche.com/shop/CategoryDisplay?catalogId=10001&tab=Assay+Design+Center&identifier=Universal+Probe+Library&langId=-1#tab-3 (accessed on 20 September 2016).
- Schmidt, M.; Finley, D. Regulation of proteasome activity in health and disease. Biochim. Biophys. Acta 2014, 1843, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bachovchin, D.A.; Cravatt, B.F. The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov. 2012, 11, 52–68. [Google Scholar] [CrossRef]
- Scaloni, A.; Jones, W.; Pospischil, M.; Sassa, S.; Schneewind, O.; Popowicz, A.M.; Bossa, F.; Graziano, S.L.; Manning, J.M. Deficiency of acylpeptide hydrolase in small-cell lung carcinoma cell lines. J. Lab. Clin. Med. 1992, 120, 546–552. [Google Scholar]
- Perrier, J.; Durand, A.; Giardina, T.; Puigserver, A. Catabolism of intracellular N-terminal acetylated proteins: Involvement of acylpeptide hydrolase and acylase. Biochimie 2005, 87, 673–685. [Google Scholar] [CrossRef]
- Yamin, R.; Zhao, C.; O’Connor, P.B.; McKee, A.C.; Abraham, C.R. Acyl peptide hydrolase degrades monomeric and oligomeric amyloid-β peptide. Mol. Neurodegener. 2009, 4, 33. [Google Scholar] [CrossRef] [PubMed]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palumbo, R.; Gogliettino, M.; Cocca, E.; Iannitti, R.; Sandomenico, A.; Ruvo, M.; Balestrieri, M.; Rossi, M.; Palmieri, G. APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome. Int. J. Mol. Sci. 2016, 17, 1614. https://doi.org/10.3390/ijms17101614
Palumbo R, Gogliettino M, Cocca E, Iannitti R, Sandomenico A, Ruvo M, Balestrieri M, Rossi M, Palmieri G. APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome. International Journal of Molecular Sciences. 2016; 17(10):1614. https://doi.org/10.3390/ijms17101614
Chicago/Turabian StylePalumbo, Rosanna, Marta Gogliettino, Ennio Cocca, Roberta Iannitti, Annamaria Sandomenico, Menotti Ruvo, Marco Balestrieri, Mosè Rossi, and Gianna Palmieri. 2016. "APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome" International Journal of Molecular Sciences 17, no. 10: 1614. https://doi.org/10.3390/ijms17101614
APA StylePalumbo, R., Gogliettino, M., Cocca, E., Iannitti, R., Sandomenico, A., Ruvo, M., Balestrieri, M., Rossi, M., & Palmieri, G. (2016). APEH Inhibition Affects Osteosarcoma Cell Viability via Downregulation of the Proteasome. International Journal of Molecular Sciences, 17(10), 1614. https://doi.org/10.3390/ijms17101614