
The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer


Abstract
:1. Introduction
2. Results
2.1. Analysis of Clinical Data
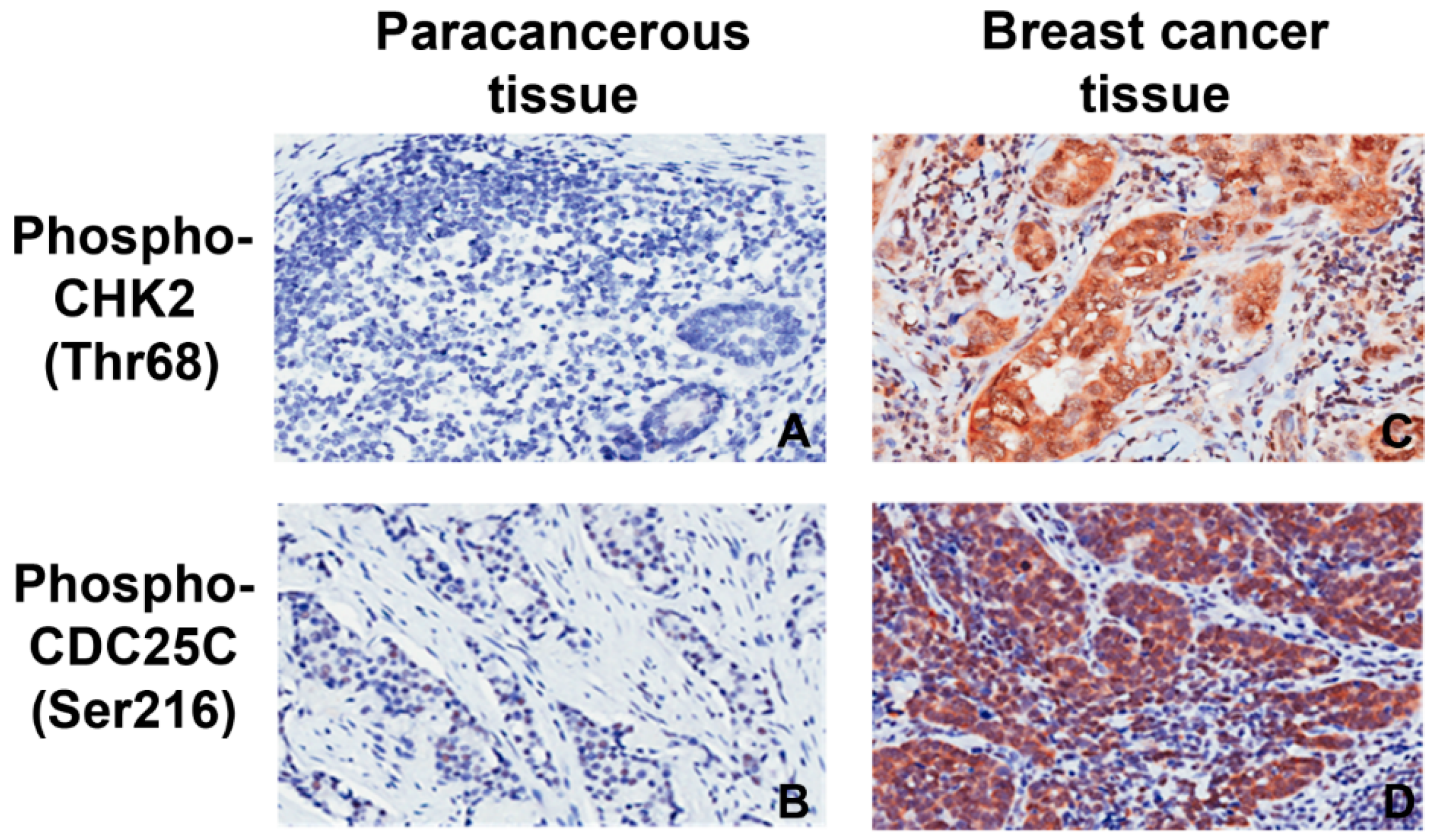
2.2. Expression of pCHK2-Thr68 and pCDC25C-Ser216
2.3. pCHK2-Thr68 and pCDC25C-Ser216 in Relation to Clinicopathological Factors
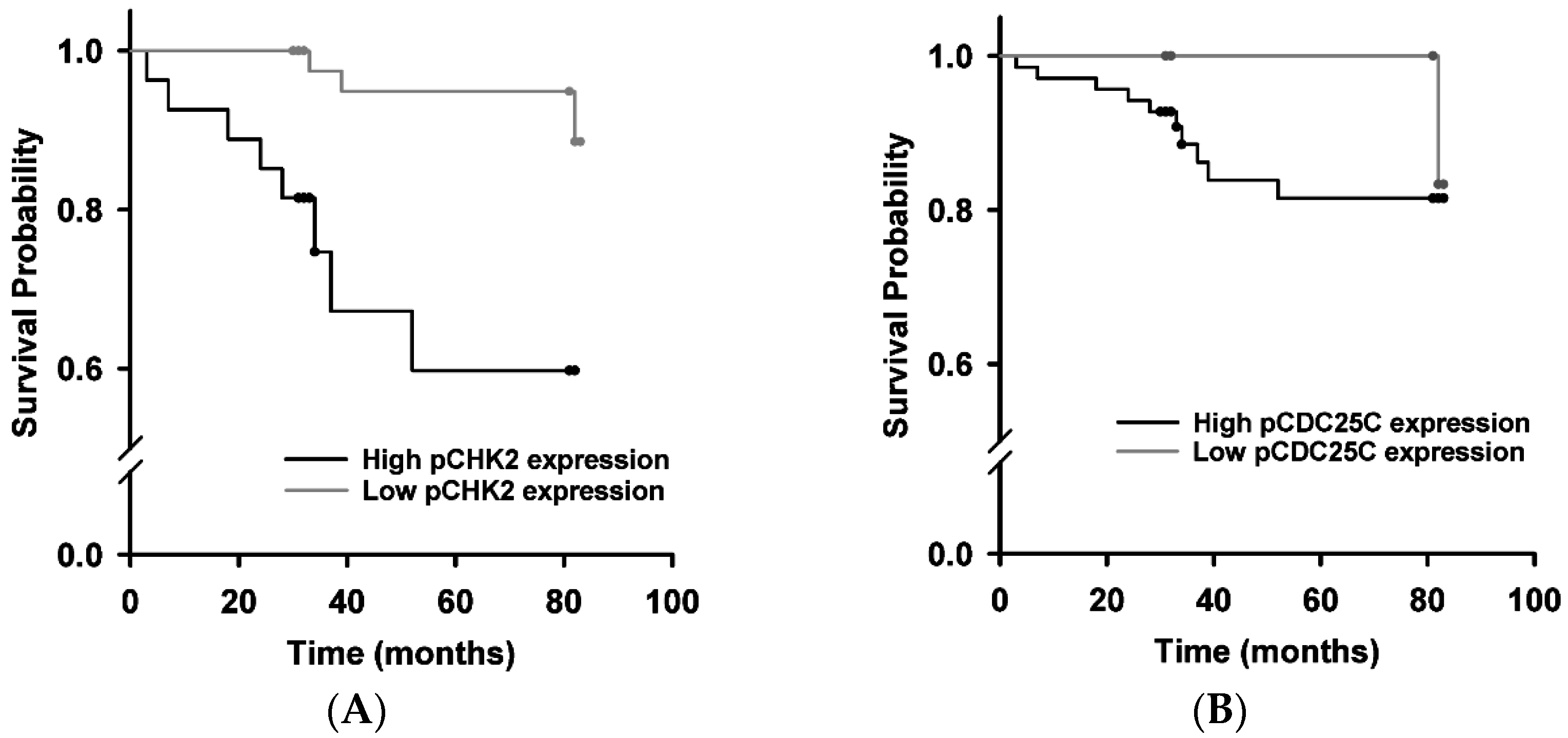
2.4. Survival Analysis
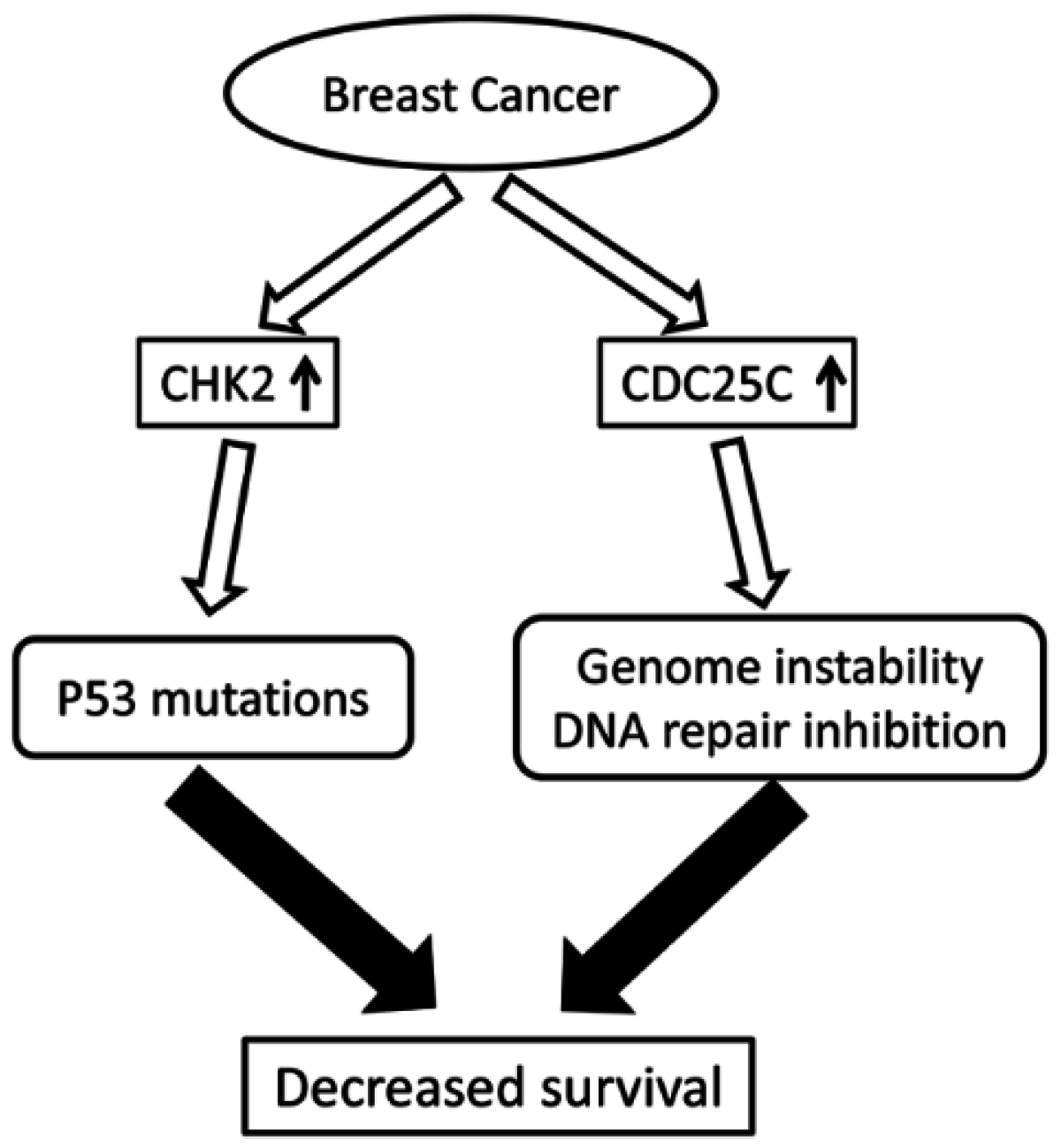
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Experimental Methods
4.2.1. Collection of Specimens
4.2.2. Tissue Chip and Immunohistochemical Staining (IHC)
4.2.3. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, S.S.; Ekwueme, D.U. Breast cancer as a global health concern. Cancer Epidemiol. 2009, 33, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J.; Bartkova, J. DNA damage response as an anti-cancer barrier: Damage threshold and the concept of “conditional haploinsufficiency”. Cell Cycle 2007, 6, 2344–2347. [Google Scholar] [CrossRef] [PubMed]
- Ralhan, R.; Kaur, J.; Kreienberg, R.; Wiesmuller, L. Links between DNA double strand break repair and breast cancer: Accumulating evidence from both familial and nonfamilial cases. Cancer Lett. 2007, 248, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Wang, N.; Wang, Y.J. XRCC3 and RAD51 expression are associated with clinical factors in breast cancer. PLoS ONE 2013, 8, e72104. [Google Scholar] [CrossRef] [PubMed]
- Boutros, R.; Lobjois, V.; Ducommun, B. CDC25 phosphatases in cancer cells: Key players? Good targets? Nat. Rev. Cancer 2007, 7, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Sur, S.; Agrawal, D.K. Phosphatases and kinases regulating CDC25 activity in the cell cycle: Clinical implications of CDC25 overexpression and potential treatment strategies. Mol. Cell. Biochem. 2016, 416, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Kristjansdottir, K.; Rudolph, J. Cdc25 phosphatases and cancer. Chem. Biol. 2004, 11, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Yoshida, H.; Uruno, T.; Takamura, Y.; Miya, A.; Kuma, K.; Miyauchi, A. Expression of cdc25A and cdc25B phosphatase in breast carcinoma. Breast Cancer 2004, 11, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Trope, C.G.; Florenes, V.A.; Suo, Z.; Nesland, J.M.; Holm, R. Overexpression of CDC25B, CDC25C and phospho-CDC25C (Ser216) in vulvar squamous cell carcinomas are associated with malignant features and aggressive cancer phenotypes. BMC Cancer 2010, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Albert, H.; Battaglia, E.; Monteiro, C.; Bagrel, D. Genotoxic stress modulates CDC25C phosphatase alternative splicing in human breast cancer cell lines. Mol. Oncol. 2012, 6, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.C.; Su, N.; Shi, X.J.; Zhao, W.; Ke, Y.; Zi, X.; Zhao, N.M.; Qin, Y.H.; Zhao, H.W.; Liu, H.M. Jaridonin-induced G2/M phase arrest in human esophageal cancer cells is caused by reactive oxygen species-dependent Cdc2-tyr15 phosphorylation via ATM-Chk1/2-Cdc25C pathway. Toxicol. Appl. Pharmacol. 2015, 282, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Kilpivaara, O.; Laiho, P.; Aaltonen, L.A.; Nevanlinna, H. CHEK2 1100delC and colorectal cancer. J. Med. Genet. 2003, 40, e110. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Weischer, M.; Bojesen, S.E.; Tybjaerg-Hansen, A.; Axelsson, C.K.; Nordestgaard, B.G. Increased risk of breast cancer associated with CHEK2*1100delC. J. Clin. Oncol. 2007, 25, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Shigeishi, H.; Yokozaki, H.; Oue, N.; Kuniyasu, H.; Kondo, T.; Ishikawa, T.; Yasui, W. Increased expression of CHK2 in human gastric carcinomas harboring p53 mutations. Int. J. Cancer 2002, 99, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Tort, F.; Hernandez, S.; Bea, S.; Martinez, A.; Esteller, M.; Herman, J.G.; Puig, X.; Camacho, E.; Sanchez, M.; Nayach, I.; et al. Chk2-decreased protein expression and infrequent genetic alterations mainly occur in aggressive types of non-hodgkin lymphomas. Blood 2002, 100, 4602–4608. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Arora, A.; Alsubhi, N.; Agarwal, D.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.Y.; Green, A.R.; Rakha, E.; et al. Clinicopathological significance of ATM-Chk2 expression in sporadic breast cancers: A comprehensive analysis in large cohorts. Neoplasia 2014, 16, 982–991. [Google Scholar] [CrossRef] [PubMed]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Guler, G.; Himmetoglu, C.; Jimenez, R.E.; Geyer, S.M.; Wang, W.P.; Costinean, S.; Pilarski, R.T.; Morrison, C.; Suren, D.; Liu, J.; et al. Aberrant expression of DNA damage response proteins is associated with breast cancer subtype and clinical features. Breast Cancer Res. Treat. 2011, 129, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, D.; Kusdra, L.; Huskey, N.E.; Chandriani, S.; Lenburg, M.E.; Gonzalez-Angulo, A.M.; Creasman, K.J.; Bazarov, A.V.; Smyth, J.W.; Davis, S.E.; et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J. Exp. Med. 2012, 209, 679–696. [Google Scholar] [CrossRef] [PubMed]
- Tarasewicz, E.; Rivas, L.; Hamdan, R.; Dokic, D.; Parimi, V.; Bernabe, B.P.; Thomas, A.; Shea, L.D.; Jeruss, J.S. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle 2014, 13, 3191–3201. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ginestier, C.; Charafe-Jauffret, E.; Foco, H.; Kleer, C.G.; Merajver, S.D.; Dontu, G.; Wicha, M.S. BRCA1 regulates human mammary stem/progenitor cell fate. Proc. Natl. Acad. Sci. USA 2008, 105, 1680–1685. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. Brca1: Cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, L.; Buscemi, G.; Fontanella, E.; Delia, D. A protein phosphatase feedback mechanism regulates the basal phosphorylation of Chk2 kinase in the absence of DNA damage. Biochim. Biophys. Acta 2010, 1803, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Oka, K.; Tanaka, T.; Enoki, T.; Yoshimura, K.; Ohshima, M.; Kubo, M.; Murakami, T.; Gondou, T.; Minami, Y.; Takemoto, Y.; et al. DNA damage signaling is activated during cancer progression in human colorectal carcinoma. Cancer Biol. Ther. 2010, 9, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Yajima, N.; Wada, R.; Matsuzaki, Y.; Yagihashi, S. DNA damage response and its clinicopathological relationship in appendiceal tumors. Int. J. Colorectal Dis. 2014, 29, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Guldberg, P.; Gronbaek, K.; Koed, K.; Primdahl, H.; Moller, K.; Lukas, J.; Orntoft, T.F.; Bartek, J. Aberrations of the Chk2 tumour suppressor in advanced urinary bladder cancer. Oncogene 2004, 23, 8545–8551. [Google Scholar] [CrossRef] [PubMed]
- Kshirsagar, M.; Jiang, W.; Shih Ie, M. DNA damage response is prominent in ovarian high-grade serous carcinomas, especially those with Rsf-1 (HBXAP) overexpression. J. Oncol. 2012, 2012, 621685. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- DiTullio, R.A., Jr.; Mochan, T.A.; Venere, M.; Bartkova, J.; Sehested, M.; Bartek, J.; Halazonetis, T.D. 53BP1 functions in an ATM-dependent checkpoint pathway that is constitutively activated in human cancer. Nat. Cell Biol. 2002, 4, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Poehlmann, A.; Roessner, A. Importance of DNA damage checkpoints in the pathogenesis of human cancers. Pathol. Res. Pract. 2010, 206, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Ke, S.; Zhou, F.; Yang, H.; Wei, Y.; Gong, J.; Mei, Z.; Wu, L.; Yu, H.; Zhou, Y. Downregulation of high mobility group box 1 modulates telomere homeostasis and increases the radiosensitivity of human breast cancer cells. Int. J. Oncol. 2015, 46, 1051–1058. [Google Scholar] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S.H.; Kao, C.L.; Chen, Y.W.; Chien, C.S.; Hung, S.C.; Lo, J.F.; Chen, Y.J.; Ku, H.H.; Hsu, M.T.; Wong, T.T. Identification of CD133-positive radioresistant cells in atypical teratoid/rhabdoid tumor. PLoS ONE 2008, 3, e2090. [Google Scholar] [CrossRef] [PubMed]
- Antoni, L.; Sodha, N.; Collins, I.; Garrett, M.D. Chk2 kinase: Cancer susceptibility and cancer therapy—Two sides of the same coin? Nat. Rev. Cancer 2007, 7, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Pannell, B.K.; Re, A.T.; Best, T.M.; Wagner, P.D. Po2 cycling protects diaphragm function during reoxygenation via ROS, Akt, ERK, and mitochondrial channels. Am. J. Physiol. Cell. Physiol. 2015, 309, C759–C766. [Google Scholar]
- Zuo, L.; Hallman, A.H.; Yousif, M.K.; Chien, M.T. Oxidative stress, respiratory muscle dysfunction, and potential therapeutics in chronic obstructive pulmonary disease. Front. Biol. 2012, 7, 506–513. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| pCHK2-Thr68 Expression | pCDC25C-Ser216 Expression | Total | |||
|---|---|---|---|---|---|
| High | Low | High | Low | ||
| Breast Cancer | n = 54 (20.4%) | n = 211 (79.6%) | n = 218 (82.3%) | n = 47 (17.7%) | n = 265 |
| Paracancerous tissues | n = 0 (0%) | n = 33 (100%) | n = 8 (24.2%) | n = 25 (75.8%) | n = 33 |
| χ2/p value | 8.213/0.004 | 53.916/0.000 | |||
| pCHK2-Thr68 Expression | Total | ||
|---|---|---|---|
| High | Low | ||
| TNBC | n = 15 (32.6%) | n = 31 (67.4%) | n = 46 |
| non-TNBC | n = 39 (17.8%) | n = 180 (82.2%) | n = 219 |
| χ2/p value | 5.13/0.023 | ||
| Characteristics | pCHK2-Thr68 (n) | pCDC25C-Ser216 (n) | ||||||
|---|---|---|---|---|---|---|---|---|
| High | Low | χ2 | p Value | High | Low | χ2 | p Value | |
| Age at diagnosis (years) | ||||||||
| ≤40 | 4 | 16 | 2.637 * | 0.282 | 5 | 15 | 1.176 * | 0.554 |
| 40–60 | 30 | 140 | 28 | 142 | ||||
| >60 | 20 | 55 | 14 | 61 | ||||
| Tumor size (cm) | ||||||||
| ≤2 | 18 | 70 | 3.977 | 0.137 | 13 | 175 | 1.156 * | 0.563 |
| 2–5 | 28 | 127 | 31 | 124 | ||||
| >5 | 8 | 14 | 3 | 19 | ||||
| Number of lymph metastases | ||||||||
| 0 | 29 | 116 | 0.534 * | 0.923 | 29 | 116 | 6.834 * | 0.071 |
| 1–3 | 14 | 53 | 7 | 60 | ||||
| 4–9 | 7 | 22 | 4 | 25 | ||||
| ≥10 | 4 | 20 | 8 | 16 | ||||
| TNM stage | ||||||||
| 1 | 10 | 47 | 0.423 | 0.809 | 12 | 45 | 1.446 | 0.485 |
| 2 | 30 | 115 | 22 | 123 | ||||
| 3 | 14 | 49 | 13 | 50 | ||||
| Pathology type | ||||||||
| IDC | 49 | 177 | 1.610 | 0.205 | 39 | 187 | 0.242 | 0.623 |
| Non-IDC | 5 | 34 | 8 | 31 | ||||
| Histology grade | ||||||||
| I | 0 | 6 | 1.507 * | 0.441 | 1 | 5 | 0.371 * | 0.885 |
| II | 37 | 150 | 32 | 155 | ||||
| III | 17 | 55 | 14 | 58 | ||||
| HER2 | ||||||||
| + | 17 | 53 | 1.096 | 0.316 | 14 | 56 | 0.896 | 0.344 |
| - | 37 | 158 | 33 | 162 | ||||
| ER | ||||||||
| + | 25 | 128 | 3.637 | 0.057 | 25 | 128 | 0.484 | 0.487 |
| - | 29 | 83 | 22 | 90 | ||||
| PR | ||||||||
| + | 27 | 129 | 2.203 | 0.138 | 24 | 132 | 1.437 | 0.231 |
| - | 27 | 82 | 23 | 86 | ||||
| Menopausal status | ||||||||
| Premenopausal | 20 | 95 | 1.117 | 0.291 | 15 | 100 | 3.066 | 0.08 |
| Postmenopausal | 34 | 116 | 32 | 118 | ||||
| Dependent Variable | Independent Variable | Regression Coefficient | SE | p Value | OR | 95% CI |
|---|---|---|---|---|---|---|
| pCHK2-Thr68 | pCDC25C-Ser216 | 0.849 | 0.312 | 0.006 | 2.337 | 1.267–4.309 |
| pCDC25C-Ser216 | pCHK2-Thr68 | 1.085 | 0.337 | 0.002 | 2.524 | 1.462–4.625 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, H.; Wang, B.; Zhang, F.; Qian, Y.; Chuang, C.-C.; Ying, M.; Wang, Y.; Zuo, L. The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer. Int. J. Mol. Sci. 2016, 17, 1803. https://doi.org/10.3390/ijms17111803
Jiang H, Wang B, Zhang F, Qian Y, Chuang C-C, Ying M, Wang Y, Zuo L. The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer. International Journal of Molecular Sciences. 2016; 17(11):1803. https://doi.org/10.3390/ijms17111803
Chicago/Turabian StyleJiang, Huayong, Bin Wang, Fuli Zhang, Yuanyu Qian, Chia-Chen Chuang, Mingzhen Ying, Yajie Wang, and Li Zuo. 2016. "The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer" International Journal of Molecular Sciences 17, no. 11: 1803. https://doi.org/10.3390/ijms17111803
APA StyleJiang, H., Wang, B., Zhang, F., Qian, Y., Chuang, C.-C., Ying, M., Wang, Y., & Zuo, L. (2016). The Expression and Clinical Outcome of pCHK2-Thr68 and pCDC25C-Ser216 in Breast Cancer. International Journal of Molecular Sciences, 17(11), 1803. https://doi.org/10.3390/ijms17111803