
The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins

Abstract
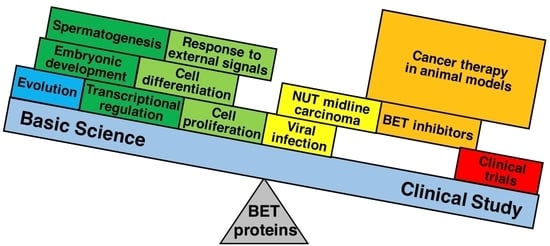
:
1. Introduction
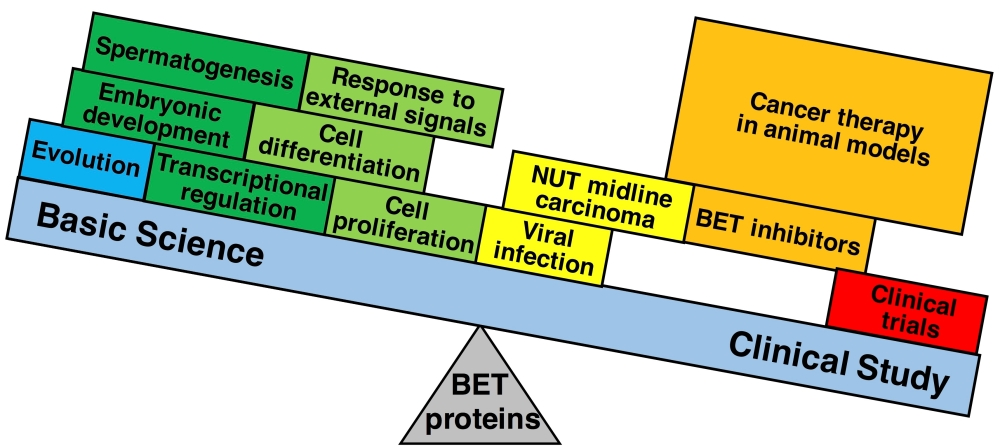
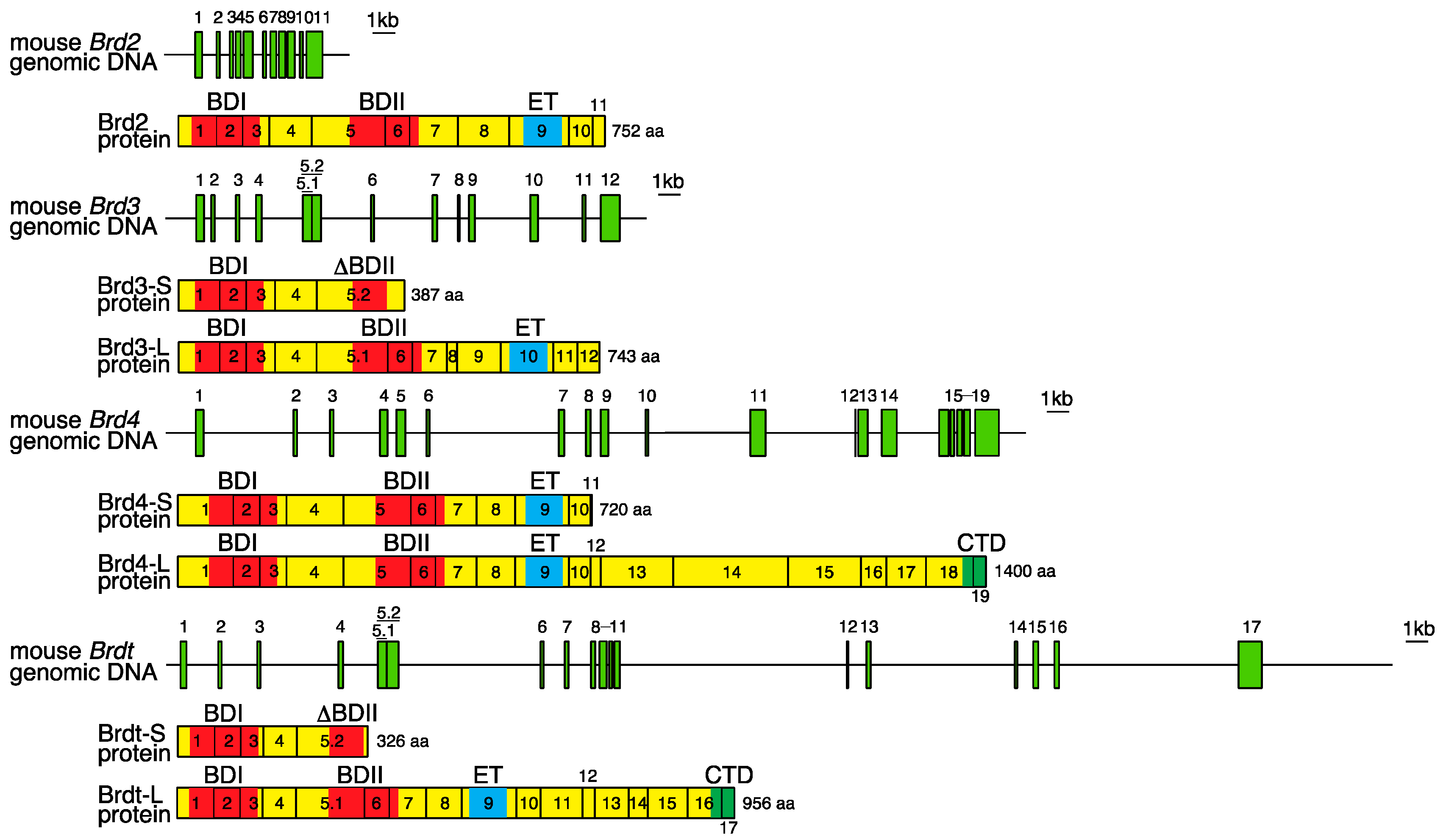
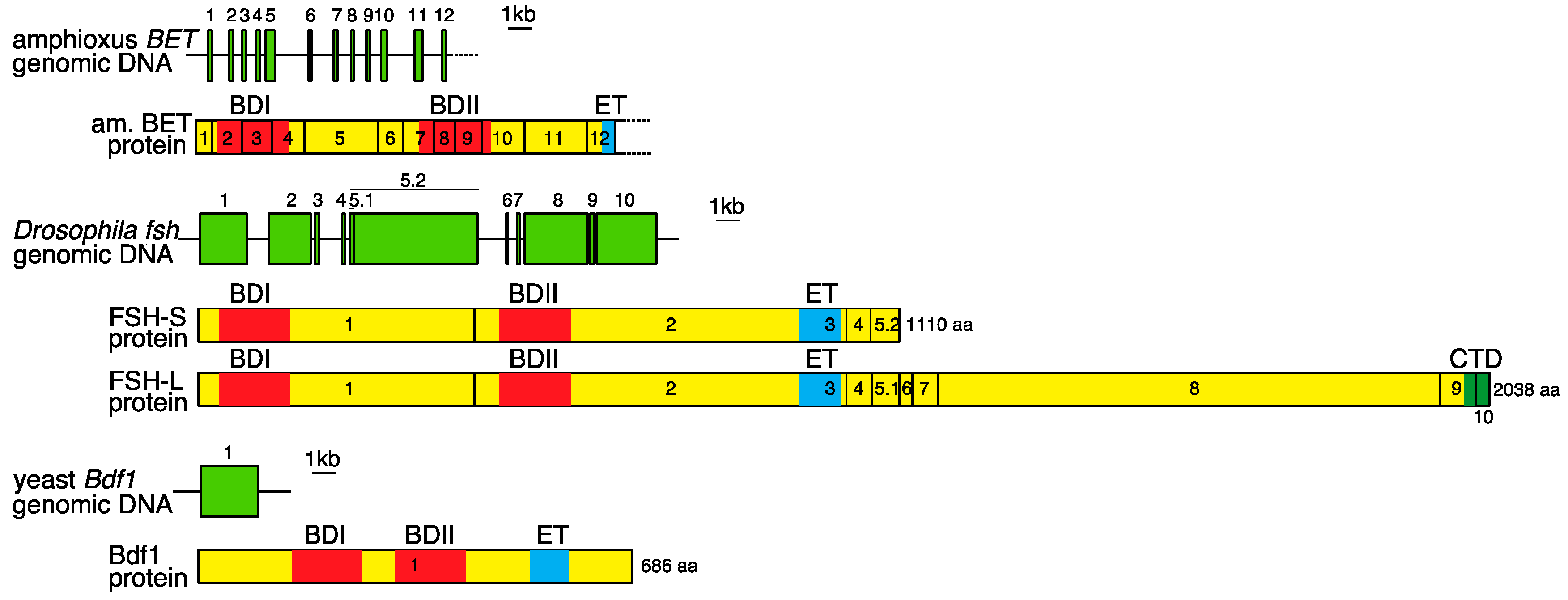
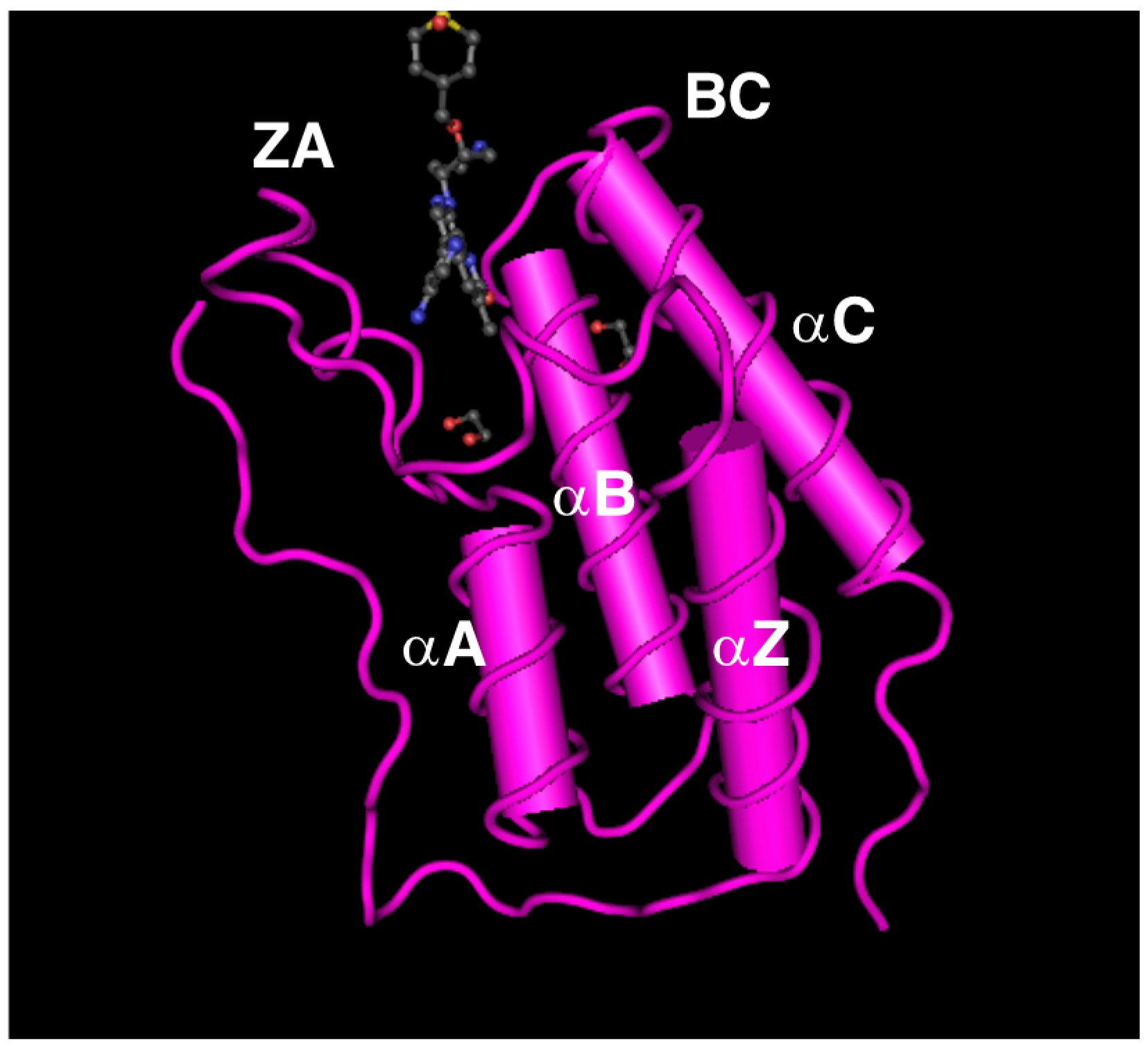
2. Genomic Organization of Bromodomain and Extra-Terminal Domain (BET) Family Genes and Structure of the Proteins Encoded by These Genes
3. Basic Functions of BRD2 and BRD3
3.1. BRD2 Plays a Role in Cell Growth and Neuronal Cell Generation
3.2. BRD2 and BRD3 Specifically Recognize Acetylated Histones through Their Bromodomains to Promote Transcription of Genes Required for Determining Cell Identities
4. Basic Functions of BRD4
4.1. BRD4 Is Required for Cellular Proliferation
4.2. BRD4 Regulates Gene Transcription by Interacting with Acetylated Histones through Its Bromodomains
4.3. BRD4 Functions in Mitotic Bookmarking
5. Basic Functions of BRDT
5.1. BRDT Is Essential for Spermatogenesis
5.2. BRDT Interacts with Various Proteins and Functions as a Transcriptional Regulator during Spermatogenesis
5.3. BRDT Exerts a Function as a Chromatin-Remodeling Factor during Spermatogenesis by Interacting with Acetylated Histones
6. Pathological Functions of BET Proteins Leading to Disease
6.1. BRD4-NUT or BRD3-NUT Fusion Protein Causes NUT Midline Carcinoma
6.2. BRD2 and BRD4 Interact with Viral Proteins and Contribute to Oncogenesis in Host Cells Infected with Viruses
6.3. BET Proteins Are Potential Therapeutic Targets in a Wide Range of Cancers
6.4. BRDT Is a Potential Target for Male Contraception
7. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Haynes, S.R.; Mozer, B.A.; Bhatia-Dey, N.; Dawid, I.B. The Drosophila fsh locus, a maternal effect homeotic gene, encodes apparent membrane proteins. Dev. Biol. 1989, 134, 246–257. [Google Scholar] [CrossRef]
- Beck, S.; Hanson, I.; Kelly, A.; Pappin, D.J.; Trowsdale, J. A homologue of the Drosophila female sterile homeotic (fsh) gene in the class II region of the human MHC. DNA Seq. 1992, 2, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Lygerou, Z.; Conesa, C.; Lesage, P.; Swanson, R.N.; Ruet, A.; Carlson, M.; Sentenac, A.; Seraphin, B. The yeast BDF1 gene encodes a transcription factor involved in the expression of a broad class of genes including snRNAs. Nucleic Acids Res. 1994, 22, 5332–5340. [Google Scholar] [CrossRef] [PubMed]
- Chua, P.; Roeder, G.S. Bdf1, a yeast chromosomal protein required for sporulation. Mol. Cell. Biol. 1995, 15, 3685–3696. [Google Scholar] [CrossRef] [PubMed]
- Haynes, S.R.; Dollard, C.; Winston, F.; Beck, S.; Trowsdale, J.; Dawid, I.B. The bromodomain: A conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Wolf, U.; Atkin, N.B. Evolution from fish to mammals by gene duplication. Hereditas 1968, 59, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, K.L.; Gorman, P.; Thomas, C.; Sheer, D.; Trowsdale, J.; Beck, S. Chromosomal localization, gene structure and transcription pattern of the ORFX gene, a homologue of the MHC-linked RING3 gene. Gene 1997, 200, 177–183. [Google Scholar] [CrossRef]
- Dey, A.; Ellenberg, J.; Farina, A.; Coleman, A.E.; Maruyama, T.; Sciortino, S.; Lippincott-Schwartz, J.; Ozato, K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. [Google Scholar]
- Jones, M.H.; Numata, M.; Shimane, M. Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics 1997, 45, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Ando, A.; Suto, Y.; Kasai, F.; Shigenari, A.; Takishima, N.; Kikkawa, E.; Iwata, K.; Kuwano, Y.; Kitamura, Y.; et al. Genomic anatomy of a premier major histocompatibility complex paralogous region on chromosome 1q21–q22. Genome Res. 2001, 11, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Abi-Rached, L.; Gilles, A.; Shiina, T.; Pontarotti, P.; Inoko, H. Evidence of en bloc duplication in vertebrate genomes. Nat. Genet. 2002, 31, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. Brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar] [CrossRef]
- Brownell, J.E.; Zhou, J.; Ranalli, T.; Kobayashi, R.; Edmondson, D.G.; Roth, S.Y.; Allis, C.D. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell 1996, 84, 843–851. [Google Scholar] [CrossRef]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. The CBP co-activator is a histone acetyltransferase. Nature 1996, 384, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar] [PubMed]
- Winston, F.; Allis, C.D. The bromodomain: A chromatin-targeting module? Nat. Struct. Biol. 1999, 6, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; Siegel, R.M.; Jang, M.K.; Lenardo, M.J.; Ozato, K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Rickards, B.; Flint, S.J. The double bromodomain proteins Brd2 and Brd3 couple histone acetylation to transcription. Mol. Cell 2008, 30, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ma, Q.; Wong, K.; Li, W.; Ohgi, K.; Zhang, J.; Aggarwal, A.K.; Rosenfeld, M.G. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 2013, 155, 1581–1595. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Kanno, Y.; LeRoy, G.; Campos, E.; Sun, H.W.; Brooks, S.R.; Vahedi, G.; Heightman, T.D.; Garcia, B.A.; Reinberg, D.; et al. BRD4 assists elongation of both coding and enhancer RNAs by interacting with acetylated histones. Nat. Struct. Mol. Biol. 2014, 21, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- LeRoy, G.; Chepelev, I.; DiMaggio, P.A.; Blanco, M.A.; Zee, B.M.; Zhao, K.; Garcia, B.A. Proteogenomic characterization and mapping of nucleosomes decoded by Brd and HP1 proteins. Genome Biol. 2012, 13, R68. [Google Scholar] [CrossRef] [PubMed]
- Gaucher, J.; Boussouar, F.; Montellier, E.; Curtet, S.; Buchou, T.; Bertrand, S.; Hery, P.; Jounier, S.; Depaux, A.; Vitte, A.L.; et al. Bromodomain-dependent stage-specific male genome programming by Brdt. EMBO J. 2012, 31, 3809–3820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, C.A.; Miyoshi, I.; Kubonishi, I.; Grier, H.E.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4-NUT fusion oncogene: A novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [Google Scholar] [PubMed]
- Alekseyenko, A.A.; Walsh, E.M.; Wang, X.; Grayson, A.R.; His, P.T.; Kharchenko, P.V.; Kuroda, M.I.; French, C.A. The oncogenic BRD4-NUT chromatin regulator drives aberrant transcription within large topological domains. Genes Dev. 2015, 29, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., III. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar] [CrossRef] [PubMed]
- Korb, E.; Herre, M.; Zucker-Scharff, I.; Darnell, R.B.; Allis, C.D. BET protein Brd4 activates transcription in neurons and BET inhibitor JQ1 blocks memory in mice. Nat. Neurosci. 2015, 18, 1464–1473. [Google Scholar] [CrossRef] [PubMed]
- Rhee, K.; Brunori, M.; Besset, V.; Trousdale, R.; Wolgemuth, D.J. Expression and potential role of Fsrg1, a murine bromodomain-containing homologue of the Drosophila gene female sterile homeotic. J. Cell Sci. 1998, 111, 3541–3550. [Google Scholar] [PubMed]
- Taniguchi, Y.; Matsuzaka, Y.; Fujimoto, H.; Miyado, K.; Kohda, A.; Okumura, K.; Kimura, M.; Inoko, H. Nucleotide sequence of the Ring3 gene in the class II region of the mouse MHC and its abundant expression in testicular germ cells. Genomics 1998, 51, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhou, M.M. Bromodomain: An acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. [Google Scholar]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Sowa, M.E.; Ottinger, M.; Smith, J.A.; Shi, Y.; Harper, J.W.; Howley, P.M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef] [PubMed]
- Schroder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnolzer, M.; Verdin, E.; et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef] [PubMed]
- Itzen, F.; Greifenberg, A.K.; Bosken, C.A.; Geyer, M. Brd4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014, 42, 7577–7590. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Suzuki, H.; Ohtsuka, M.; Kikuchi, N.; Kimura, M.; Inoko, H. Isolation and characterization of three genes paralogous to mouse Ring3. Nucleic Acids Res. Suppl. 2001, 1, 247–248. [Google Scholar] [CrossRef]
- Shang, E.; Salazar, G.; Crowley, T.E.; Wang, X.; Lopez, R.A.; Wang, X.; Wolgemuth, D.J. Identification of unique, differentiation stage-specific patterns of expression of the bromodomain-containing genes Brd2, Brd3, Brd4, and Brdt in the mouse testis. Gene Expr. Patterns 2004, 4, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Green, M.R. A novel, mitogen-activated nuclear kinase is related to a Drosophila developmental regulator. Genes Dev. 1996, 10, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.L.; King, B.; Lin, S.C.; Kennison, J.A.; Huang, D.H. A double-bromodomain protein, FSH-S, activates the homeotic gene ultrabithorax through a critical promoter-proximal region. Mol. Cell. Biol. 2007, 27, 5486–5498. [Google Scholar] [CrossRef] [PubMed]
- Denis, G.V.; Vaziri, C.; Guo, N.; Faller, D.V. RING3 kinase transactivates promoters of cell cycle regulatory genes through E2F. Cell Growth Differ. 2000, 11, 417–424. [Google Scholar] [PubMed]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Wang, X.; Wen, D.; Greenberg, D.A.; Wolgemuth, D.J. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev. Dyn. 2009, 238, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Gyuris, A.; Donovan, D.J.; Seymour, K.A.; Lovasco, L.A.; Smilowitz, N.R.; Halperin, A.L.; Klysik, J.E.; Freiman, R.N. The chromatin-targeting protein Brd2 is required for neural tube closure and embryogenesis. Biochim. Biophys. Acta 2009, 1789, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Pal, D.K.; Evgrafov, O.V.; Tabares, P.; Zhang, F.; Durner, M.; Greenberg, D.A. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am. J. Hum. Genet. 2003, 73, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Velisek, L.; Shang, E.; Veliskova, J.; Chachua, T.; Macchiarulo, S.; Maglakelidze, G.; Wolgemuth, D.J.; Greenberg, D.A. GABAergic neuron deficit as an idiopathic generalized epilepsy mechanism: The role of BRD2 haploinsufficiency in juvenile myoclonic epilepsy. PLoS ONE 2011, 6, e23656. [Google Scholar] [CrossRef] [PubMed]
- Hochreiter, B.; Garcia, A.P.; Schmid, J.A. Fluorescent proteins as genetically encoded FRET biosensors in life sciences. Sensors 2015, 15, 26281–26314. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.M.; Chan, F.K.; Zacharias, D.A.; Swofford, R.; Holmes, K.L.; Tsien, R.Y.; Lenardo, M.J. Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci. STKE 2000, 38. [Google Scholar] [CrossRef] [PubMed]
- Chan, F.K.; Siegel, R.M.; Zacharias, D.; Swofford, R.; Holmes, K.L.; Tsien, R.Y.; Lenardo, M.J. Fluorescence resonance energy transfer analysis of cell surface receptor interactions and signaling using spectral variants of the green fluorescent protein. Cytometry 2001, 44, 361–368. [Google Scholar] [CrossRef]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y. Hox transcription factors: Modulators of cell–cell and cell-extracellular matrix adhesion. BioMed Res. Int. 2014, 2014, 591374. [Google Scholar] [CrossRef] [PubMed]
- Rezsohazy, R.; Saurin, A.J.; Maurel-Zaffran, C.; Graba, Y. Cellular and molecular insights into Hox protein action. Development 2015, 142, 1212–1227. [Google Scholar] [CrossRef] [PubMed]
- Digan, M.E.; Haynes, S.R.; Mozer, B.A.; Dawid, I.B.; Forquignon, F.; Gans, M. Genetic and molecular analysis of fs(1)h, a maternal effect homeotic gene in Drosophila. Dev. Biol. 1986, 114, 161–169. [Google Scholar] [CrossRef]
- Lamonica, J.M.; Deng, W.; Kadauke, S.; Campbell, A.E.; Gamsjaeger, R.; Wang, H.; Cheng, Y.; Billin, A.N.; Hardison, R.C.; Mackay, J.P.; et al. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. USA 2011, 108, E159–E168. [Google Scholar] [CrossRef] [PubMed]
- Gamsjaeger, R.; Webb, S.R.; Lamonica, J.M.; Billin, A.; Blobel, G.A.; Mackay, J.P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell. Biol. 2011, 31, 2632–2640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhang, X.; Iwama, A.; Yu, C.; Smith, K.A.; Mueller, B.U.; Narravula, S.; Torbett, B.E.; Orkin, S.H.; Tenen, D.G. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood 2000, 96, 2641–2648. [Google Scholar] [PubMed]
- Stonestrom, A.J.; Hsu, S.C.; Jahn, K.S.; Huang, P.; Keller, C.A.; Giardine, B.M.; Kadauke, S.; Campbell, A.E.; Evans, P.; Hardison, R.C.; et al. Functions of BET proteins in erythroid gene expression. Blood 2015, 125, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Phair, R.D.; Misteli, T. High mobility of proteins in the mammalian cell nucleus. Nature 2000, 404, 604–609. [Google Scholar] [PubMed]
- Phair, R.D.; Misteli, T. Kinetic modelling approaches to in vivo imaging. Nat. Rev. Mol. Cell Biol. 2001, 2, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Houzelstein, D.; Bullock, S.L.; Lynch, D.E.; Grigorieva, E.F.; Wilson, V.A.; Beddington, R.S. Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 2002, 22, 3794–3802. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Stein, P.; Cheng, X.; Yang, W.; Shao, N.Y.; Morrisey, E.E.; Schultz, R.M.; You, J. BRD4 regulates Nanog expression in mouse embryonic stem cells and preimplantation embryos. Cell Death Differ. 2014, 21, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Watanabe, D.; Handa, H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998, 17, 7395–7403. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Yamada, T.; Yamaguchi, Y.; Inukai, N.; Okamoto, S.; Mura, T.; Handa, H. P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol. Cell 2006, 21, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yang, Z.; Zhou, Q. Phosphorylated positive transcription elongation factor b (P-TEFb) is tagged for inhibition through association with 7SK snRNA. J. Biol. Chem. 2004, 279, 4153–4160. [Google Scholar] [CrossRef] [PubMed]
- Yik, J.H.; Chen, R.; Nishimura, R.; Jennings, J.L.; Link, A.J.; Zhou, Q. Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 2003, 12, 971–982. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J., III; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed]
- Angrand, P.O.; Apiou, F.; Stewart, A.F.; Dutrillaux, B.; Losson, R.; Chambon, P. NSD3, a new SET domain-containing gene, maps to 8p12 and is amplified in human breast cancer cell lines. Genomics 2001, 74, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Heintzman, N.D.; Ren, B. Finding distal regulatory elements in the human genome. Curr. Opin. Genet. Dev. 2009, 19, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.C.; Debrosse, M.; Smith, M.; Dey, A.; Huynh, W.; Sarai, N.; Heightman, T.D.; Tamura, T.; Ozato, K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol. 2013, 33, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Frey, U.; Frey, S.; Schollmeier, F.; Krug, M. Influence of actinomycin D, a RNA synthesis inhibitor, on long-term potentiation in rat hippocampal neurons in vivo and in vitro. J. Physiol. 1996, 490, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, E.; Ying, S.W.; Kanhema, T.; Croll, S.D.; Bramham, C.R. Brain-derived neurotrophic factor triggers transcription-dependent, late phase long-term potentiation in vivo. J. Neurosci. 2002, 22, 7453–7461. [Google Scholar] [PubMed]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Caceres, J.F. Cellular stress and RNA splicing. Trends Biochem. Sci. 2009, 34, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Hussong, M.; Borno, S.T.; Kerick, M.; Wunderlich, A.; Franz, A.; Sultmann, H.; Timmermann, B.; Lehrach, H.; Hirsch-Kauffmann, M.; Schweiger, M.R. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014, 5, e1195. [Google Scholar] [CrossRef] [PubMed]
- Hussong, M.; Kaehler, C.; Kerick, M.; Grimm, C.; Franz, A.; Timmermann, B.; Welzel, F.; Isensee, J.; Hucho, T.; Krobitsch, S.; et al. The bromodomain protein BRD4 regulates splicing during heat shock. Nucleic Acids Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef]
- Phair, R.D.; Scaffidi, P.; Elbi, C.; Vecerova, J.; Dey, A.; Ozato, K.; Brown, D.T.; Hager, G.; Bustin, M.; Misteli, T. Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 2004, 24, 6393–6402. [Google Scholar] [CrossRef] [PubMed]
- Sprague, B.L.; McNally, J.G. FRAP analysis of binding: Proper and fitting. Trends Cell Biol. 2005, 15, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Nishiyama, A.; Karpova, T.; McNally, J.; Ozato, K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, E.F.; Sanford, S.; Levens, D. Marking of active genes on mitotic chromosomes. Nature 1997, 388, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Nakamura, T.; Fu, Y.; Lazar, Z.; Spector, D.L. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nat. Cell Biol. 2011, 13, 1295–1304. [Google Scholar] [CrossRef] [PubMed]
- Janicki, S.M.; Tsukamoto, T.; Salghetti, S.E.; Tansey, W.P.; Sachidanandam, R.; Prasanth, K.V.; Ried, T.; Shav-Tal, Y.; Bertrand, E.; Singer, R.H.; et al. From silencing to gene expression: Real-time analysis in single cells. Cell 2004, 116, 683–698. [Google Scholar] [CrossRef]
- Kumaran, R.I.; Spector, D.L. A genetic locus targeted to the nuclear periphery in living cells maintains its transcriptional competence. J. Cell Biol. 2008, 180, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Shang, E.; Nickerson, H.D.; Wen, D.; Wang, X.; Wolgemuth, D.J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Matzuk, M.M.; Sung, W.K.; Guo, Q.; Wang, P.; Wolgemuth, D.J. Cyclin A1 is required for meiosis in the male mouse. Nat. Genet. 1998, 20, 377–380. [Google Scholar] [PubMed]
- Nickerson, H.D.; Joshi, A.; Wolgemuth, D.J. Cyclin A1-deficient mice lack histone H3 serine 10 phosphorylation and exhibit altered aurora B dynamics in late prophase of male meiosis. Dev. Biol. 2007, 306, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Dhar, S.; Thota, A.; Rao, M.R. Insights into role of bromodomain, testis-specific (Brdt) in acetylated histone H4-dependent chromatin remodeling in mammalian spermiogenesis. J. Biol. Chem. 2012, 287, 6387–6405. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Wang, L.; Guarnieri, P.; Wolgemuth, D.J. The testis-specific double bromodomain-containing protein BRDT forms a complex with multiple spliceosome components and is required for mRNA splicing and 3′-UTR truncation in round spermatids. Nucleic Acids Res. 2012, 40, 7162–7175. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wolgemuth, D.J. BET protein BRDT complexes with HDAC1, PRMT5, and TRIM28 and functions in transcriptional repression during spermatogenesis. J. Cell. Biochem. 2016, 117, 1429–1438. [Google Scholar] [CrossRef] [PubMed]
- Pivot-Pajot, C.; Caron, C.; Govin, J.; Vion, A.; Rousseaux, S.; Khochbin, S. Acetylation-dependent chromatin reorganization by BRDT, a testis-specific bromodomain-containing protein. Mol. Cell. Biol. 2003, 23, 5354–5365. [Google Scholar] [CrossRef] [PubMed]
- Pierce, M.M.; Raman, C.S.; Nall, B.T. Isothermal titration calorimetry of protein–protein interactions. Methods 1999, 19, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Moriniere, J.; Rousseaux, S.; Steuerwald, U.; Soler-Lopez, M.; Curtet, S.; Vitte, A.L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Ito, T.; Nishino, N.; Khochbin, S.; Yoshida, M. Real-time imaging of histone H4 hyperacetylation in living cells. Proc. Natl. Acad. Sci. USA 2009, 106, 16257–16262. [Google Scholar] [CrossRef] [PubMed]
- Rathke, C.; Baarends, W.M.; Awe, S.; Renkawitz-Pohl, R. Chromatin dynamics during spermiogenesis. Biochim. Biophys. Acta 2014, 1839, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Meistrich, M.L.; Trostle-Weige, P.K.; Lin, R.; Bhatnagar, Y.M.; Allis, C.D. Highly acetylated H4 is associated with histone displacement in rat spermatids. Mol. Reprod. Dev. 1992, 31, 170–181. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Miyoshi, I.; Aster, J.C.; Kubonishi, I.; Kroll, T.G.; Dal Cin, P.; Vargas, S.O.; Perez-Atayde, A.R.; Fletcher, J.A. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19). Am. J. Pathol. 2001, 159, 1987–1992. [Google Scholar] [CrossRef]
- Ball, A.; Bromley, A.; Glaze, S.; French, C.A.; Ghatage, P.; Kobel, M. A rare case of NUT midline carcinoma. Gynecol. Oncol. Case Rep. 2012, 3, 1–3. [Google Scholar] [CrossRef] [PubMed]
- French, C.A.; Ramirez, C.L.; Kolmakova, J.; Hickman, T.T.; Cameron, M.J.; Thyne, M.E.; Kutok, J.L.; Toretsky, J.A.; Tadavarthy, A.K.; Kees, U.R.; et al. BRD-NUT oncoproteins: A family of closely related nuclear proteins that block epithelial differentiation and maintain the growth of carcinoma cells. Oncogene 2008, 27, 2237–2242. [Google Scholar] [CrossRef] [PubMed]
- Haruki, N.; Kawaguchi, K.S.; Eichenberger, S.; Massion, P.P.; Gonzalez, A.; Gazdar, A.F.; Minna, J.D.; Carbone, D.P.; Dang, T.P. Cloned fusion product from a rare t(15;19)(q13.2;p13.1) inhibit S phase in vitro. J. Med. Genet. 2005, 42, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Diaz, J.; Jiao, J.; Wang, R.; You, J. Perturbation of BRD4 protein function by BRD4-NUT protein abrogates cellular differentiation in NUT midline carcinoma. J. Biol. Chem. 2011, 286, 27663–27675. [Google Scholar] [CrossRef] [PubMed]
- Reynoird, N.; Schwartz, B.E.; Delvecchio, M.; Sadoul, K.; Meyers, D.; Mukherjee, C.; Caron, C.; Kimura, H.; Rousseaux, S.; Cole, P.A.; et al. Oncogenesis by sequestration of CBP/p300 in transcriptionally inactive hyperacetylated chromatin domains. EMBO J. 2010, 29, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Alekseyenko, A.A.; Gorchakov, A.A.; Kharchenko, P.V.; Kuroda, M.I. Reciprocal interactions of human C10orf12 and C17orf96 with PRC2 revealed by BioTAP-XL cross-linking and affinity purification. Proc. Natl. Acad. Sci. USA 2014, 111, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Platt, G.M.; Simpson, G.R.; Mittnacht, S.; Schulz, T.F. Latent nuclear antigen of Kaposi’s sarcoma-associated herpesvirus interacts with RING3, a homolog of the Drosophila female sterile homeotic (fsh) gene. J. Virol. 1999, 73, 9789–9795. [Google Scholar] [PubMed]
- Viejo-Borbolla, A.; Ottinger, M.; Bruning, E.; Burger, A.; Konig, R.; Kati, E.; Sheldon, J.A.; Schulz, T.F. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 2005, 79, 13618–13629. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Srinivasan, V.; Denis, G.V.; Harrington, W.J., Jr.; Ballestas, M.E.; Kaye, K.M.; Howley, P.M. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol. 2006, 80, 8909–8919. [Google Scholar] [CrossRef] [PubMed]
- Ottinger, M.; Christalla, T.; Nathan, K.; Brinkmann, M.M.; Viejo-Borbolla, A.; Schulz, T.F. Kaposi’s sarcoma-associated herpesvirus LANA-1 interacts with the short variant of BRD4 and releases cells from a BRD4- and BRD2/RING3-induced G1 cell cycle arrest. J. Virol. 2006, 80, 10772–10786. [Google Scholar] [CrossRef] [PubMed]
- Bastien, N.; McBride, A.A. Interaction of the papillomavirus E2 protein with mitotic chromosomes. Virology 2000, 270, 124–134. [Google Scholar]
- Howley, P.M.; Lowy, D.R. Papillomaviruses and their replication. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; p. 2197. [Google Scholar]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Thierry, F.; Howley, P.M. Functional analysis of E2-mediated repression of the HPV18 P105 promoter. New Biol. 1991, 3, 90–100. [Google Scholar] [PubMed]
- You, J.; Croyle, J.L.; Nishimura, A.; Ozato, K.; Howley, P.M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. [Google Scholar] [CrossRef]
- Wu, S.Y.; Lee, A.Y.; Hou, S.Y.; Kemper, J.K.; Erdjument-Bromage, H.; Tempst, P.; Chiang, C.M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, M.R.; You, J.; Howley, P.M. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 2006, 80, 4276–4285. [Google Scholar] [CrossRef] [PubMed]
- McPhillips, M.G.; Oliveira, J.G.; Spindler, J.E.; Mitra, R.; McBride, A.A. Brd4 is required for E2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 2006, 80, 9530–9543. [Google Scholar] [CrossRef] [PubMed]
- Helfer, C.M.; Yan, J.; You, J. The cellular bromodomain protein Brd4 has multiple functions in E2-mediated papillomavirus transcription activation. Viruses 2014, 6, 3228–3249. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Henssen, A.; Thor, T.; Odersky, A.; Heukamp, L.; El-Hindy, N.; Beckers, A.; Speleman, F.; Althoff, K.; Schafers, S.; Schramm, A.; et al. BET bromodomain protein inhibition is a therapeutic option for medulloblastoma. Oncotarget 2013, 4, 2080–2095. [Google Scholar] [CrossRef] [PubMed]
- Puissant, A.; Frumm, S.M.; Alexe, G.; Bassil, C.F.; Qi, J.; Chanthery, Y.H.; Nekritz, E.A.; Zeid, R.; Gustafson, W.C.; Greninger, P.; et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013, 3, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Jacques, C.; Lamoureux, F.; Baud’huin, M.; Rodriguez Calleja, L.; Quillard, T.; Amiaud, J.; Tirode, F.; Redini, F.; Bradner, J.E.; Heymann, D.; et al. Targeting the epigenetic readers in Ewing sarcoma inhibits the oncogenic transcription factor EWS/Fli1. Oncotarget 2016, 7, 24125–24140. [Google Scholar] [CrossRef] [PubMed]
- Togel, L.; Nightingale, R.; Chueh, A.C.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.M.; Arango, D.; Dhillon, A.S.; et al. Dual targeting of bromodomain and extraterminal domain proteins, and WNT or MAPK signaling, inhibits c-MYC expression and proliferation of colorectal cancer cells. Mol. Cancer Ther. 2016, 15, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.R. Chemical probes: A shared toolbox. Nature 2016, 533, S60–S61. [Google Scholar] [CrossRef] [PubMed]
- Fong, C.Y.; Gilan, O.; Lam, E.Y.; Rubin, A.F.; Ftouni, S.; Tyler, D.; Stanley, K.; Sinha, D.; Yeh, P.; Morison, J.; et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Rathert, P.; Roth, M.; Neumann, T.; Muerdter, F.; Roe, J.S.; Muhar, M.; Deswal, S.; Cerny-Reiterer, S.; Peter, B.; Jude, J.; et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Matzuk, M.M.; McKeown, M.R.; Filippakopoulos, P.; Li, Q.; Ma, L.; Agno, J.E.; Lemieux, M.E.; Picaud, S.; Yu, R.N.; Qi, J.; et al. Small-molecule inhibition of BRDT for male contraception. Cell 2012, 150, 673–684. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BET Protein | Functions | References |
|---|---|---|
| BRD2 | • Promotion of E2F-dependent cell cycle progression in HeLa and HEK293 cells | [47,48] |
| • Closure of the neural tube in mouse embryos | [49,50] | |
| • Maintenance of the number of GABAergic neurons in the neocortex and the striatum of mice | [52] | |
| • Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | [23] | |
| • Transcriptional activation of HOXA11and D11 in HEK293 cells | [26] | |
| • Enhancement of GATA1-mediated erythroid gene activation | [63] | |
| • Interaction with LANA of KSHV that mediates episomal replication and persistence of viral genomes | [112,113] | |
| BRD3 | • Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | [23] |
| • Transcriptional activation of HOXB3, B4, B5, B6, C8, C9, C10, A3, A5, A6, and A7 in HEK293 cells | [26] | |
| • Enhancement of GATA1-mediated erythroid gene activation | [63] | |
| • Carcinogenesis induced by BRD3-NUT fusion protein | [106] | |
| BRD4 | • Stimulation of G2/M transition in HeLa cells | [8] |
| • Cell cycle progression in P19 embryonic carcinoma cells | [19] | |
| • Maintenance of inner cell mass in mouse blastocysts | [66] | |
| • Transcriptional activation of Nanog required for maintaining the pluripotency of ES cells | [67] | |
| • Release from a pause in transcription elongation | [21,24] | |
| • Assist of transcription in hyperacetylated chromatin (Property of histone-chaperone) | [25] | |
| • Transcriptional activation of c-Myc and Klf4 in NIH3T3 cells | [25] | |
| • Transcriptional activation of HOXB2, B3, B4, B5, B6, B7, B8, A4, and C5 in HEK293 cells | [26] | |
| • Transcriptional regulation of genes involved in learning and memory in mice | [34] | |
| • Enhancement of INF-induced gene transcription | [77] | |
| • Signal transducer of the cellular response to oxidative stress | [82] | |
| • Prevention of splicing inhibition in heat stress-induced cells | [83] | |
| • A gene bookmark for transcriptional reactivation in post-mitotic cells | [87,89] | |
| • Carcinogenesis induced by BRD4-NUT fusion protein | [28,106] | |
| • Interaction with LANA of KSHV that mediates episomal replication and persistence of viral genomes | [114,115] | |
| • Tethering of BPV genome to host mitotic chromosomes | [121] | |
| • Transcriptional regulation of E2 that mediates episomal maintenance and DNA replication of HPV genome | [122,124,125] | |
| BRDT | • Transcriptional regulation of genes responsible for meiotic progression during spermatogenesis | [27] |
| • Splicing machinery in testicular cells | [96] | |
| • Chromatin remodeling in MEL, 3T3, and COS7 cells | [95,98,101] | |
| • Histone replacement at post-meiotic stages during spermatogenesis | [27] |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. https://doi.org/10.3390/ijms17111849
Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. International Journal of Molecular Sciences. 2016; 17(11):1849. https://doi.org/10.3390/ijms17111849
Chicago/Turabian StyleTaniguchi, Yasushi. 2016. "The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins" International Journal of Molecular Sciences 17, no. 11: 1849. https://doi.org/10.3390/ijms17111849
APA StyleTaniguchi, Y. (2016). The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. International Journal of Molecular Sciences, 17(11), 1849. https://doi.org/10.3390/ijms17111849