
Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets

 ,
, 

Abstract
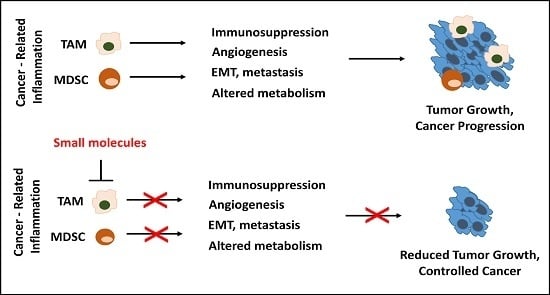
:
1. Introduction
2. Linking Inflammation and Cancer
3. Pro-Tumoral Functions and Mediators of Inflammatory Myeloid Cells, as Potential Therapeutic Targets
3.1. Immunosuppression
3.2. Angiogenesis
3.3. Epithelial-Mesenchymal Transition (EMT), Matrix Remodeling, Metastasis
3.4. Altered Metabolism
4. Therapeutic Interventions Targeting TAMs and MDSCs, Tuning the Balance
4.1. Inhibition of the Recruitment and/or Proliferation of Tumor-Associated Macrophages (TAMs) and Myeloid-Derived Suppressor Cells (MDSCs)
4.2. Selective Ablation, Depletion of TAMs and MDSCs
4.3. Re-Education of TAMs and MDSCs to Exert Anti-Tumor Functions
4.4. Differentiation of MDSCs
4.5. Pharmacological Targeting of the Pro-Tumoral Mediators of TAMs and MDSCs
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Larghi, P.; Rimoldi, M.; Totaro, M.G.; Allavena, P.; Mantovani, A.; Sica, A. Cellular and molecular pathways linking inflammation and cancer. Immunobiology 2009, 214, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Goossens, N.; Hoshida, Y. Hepatitis C virus-induced hepatocellular carcinoma. Clin. Mol. Hepatol. 2015, 21, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Beaugerie, L.; Itzkowitz, S.H. Cancers complicating inflammatory bowel disease. N. Engl. J. Med. 2015, 372, 1441–1452. [Google Scholar] [PubMed]
- Heinrich, E.L.; Walser, T.C.; Krysan, K.; Liclican, E.L.; Grant, J.L.; Rodriguez, N.L.; Dubinett, S.M. The inflammatory tumor microenvironment, epithelial mesenchymal transition and lung carcinogenesis. Cancer Microenviron. 2012, 5, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rivera, Z.; Jube, S.; Nasu, M.; Bertino, P.; Goparaju, C.; Franzoso, G.; Lotze, M.T.; Krausz, T.; Pass, H.I.; et al. Programmed necrosis induced by asbestos in human mesothelial cells causes high-mobility group box 1 protein release and resultant inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 12611–12616. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Carmi, Y.; Dotan, S.; Rider, P.; Kaplanov, I.; White, M.R.; Baron, R.; Abutbul, S.; Huszar, M.; Dinarello, C.A.; Apte, R.N.; et al. The role of IL-1β in the early tumor cell-induced angiogenic response. J. Immunol. 2013, 190, 3500–3509. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1β induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Katanov, C.; Lerrer, S.; Liubomirski, Y.; Leider-Trejo, L.; Meshel, T.; Bar, J.; Feniger-Barish, R.; Kamer, I.; Soria-Artzi, G.; Kahani, H.; et al. Regulation of the inflammatory profile of stromal cells in human breast cancer: Prominent roles for TNF-alpha and the NF-κB pathway. Stem Cell Res. Ther. 2015, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Inflammatory cytokines in cancer: Tumour necrosis factor and interleukin 6 take the stage. Ann. Rheum. Dis. 2011, 70, i104–i108. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.J.; Davidson, N.; Kuhn, R.; Muller, W.; Menon, S.; Holland, G.; Thompson-Snipes, L.; Leach, M.W.; Rennick, D. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4+ th1-like responses. J. Clin. Investig. 1996, 98, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.E.; Rao, V.P.; Poutahidis, T.; Ihrig, M.M.; Ge, Z.; Feng, Y.; Tomczak, M.; Rogers, A.B.; Horwitz, B.H.; Fox, J.G. CD4+CD25+ regulatory lymphocytes require interleukin 10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003, 63, 6042–6050. [Google Scholar] [PubMed]
- Tili, E.; Croce, C.M.; Michaille, J.J. miR-155: On the crosstalk between inflammation and cancer. Int. Rev. Immunol. 2009, 28, 264–284. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.J.; Wernicke, D.; Alder, H.; Costinean, S.; Volinia, S.; Croce, C.M. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 4908–4913. [Google Scholar] [CrossRef] [PubMed]
- Valeri, N.; Gasparini, P.; Fabbri, M.; Braconi, C.; Veronese, A.; Lovat, F.; Adair, B.; Vannini, I.; Fanini, F.; Bottoni, A.; et al. Modulation of mismatch repair and genomic stability by miR-155. Proc. Natl. Acad. Sci. USA 2010, 107, 6982–6987. [Google Scholar] [CrossRef] [PubMed]
- Kamp, D.W.; Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer: The role of the mitochondria. Oncology 2011, 25, 400–410, 413. [Google Scholar] [PubMed]
- Hiraku, Y. Formation of 8-nitroguanine, a nitrative DNA lesion, in inflammation-related carcinogenesis and its significance. Environ. Health Prev. Med. 2010, 15, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gungor, N.; Knaapen, A.M.; Munnia, A.; Peluso, M.; Haenen, G.R.; Chiu, R.K.; Godschalk, R.W.; van Schooten, F.J. Genotoxic effects of neutrophils and hypochlorous acid. Mutagenesis 2010, 25, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Marengo, B.; Nitti, M.; Furfaro, A.L.; Colla, R.; Ciucis, C.D.; Marinari, U.M.; Pronzato, M.A.; Traverso, N.; Domenicotti, C. Redox homeostasis and cellular antioxidant systems: Crucial players in cancer growth and therapy. Oxid. Med. Cell. Longev. 2016, 2016, 6235641. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [PubMed]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, inflammation, and cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef] [PubMed]
- Umar, A.; Steele, V.E.; Menter, D.G.; Hawk, E.T. Mechanisms of nonsteroidal anti-inflammatory drugs in cancer prevention. Semin. Oncol. 2016, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Allaj, V.; Guo, C.; Nie, D. Non-steroid anti-inflammatory drugs, prostaglandins, and cancer. Cell Biosci. 2013, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Riboldi, E.; Ippolito, A.; Sica, A. Molecular and epigenetic basis of macrophage polarized activation. Semin. Immunol. 2015, 27, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Katoh, H.; Watanabe, M. Myeloid-derived suppressor cells and therapeutic strategies in cancer. Mediat. Inflamm. 2015, 2015, 159269. [Google Scholar] [CrossRef] [PubMed]
- Shipp, C.; Speigl, L.; Janssen, N.; Martens, A.; Pawelec, G. A clinical and biological perspective of human myeloid-derived suppressor cells in cancer. Cell. Mol. Life Sci. 2016, 73, 4043–4061. [Google Scholar] [CrossRef] [PubMed]
- Turley, S.J.; Cremasco, V.; Astarita, J.L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 2015, 15, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Bae, J.S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.A.; Li, M.O. Ontogeny of tumor-associated macrophages and its implication in cancer regulation. Trends Cancer 2016, 2, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Caronni, N.; Savino, B.; Bonecchi, R. Myeloid cells in cancer-related inflammation. Immunobiology 2015, 220, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, T.; Shao, S.; Shi, B.; Zhao, Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology 2016, 5, e1004983. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Varner, J.A. Myeloid cells in tumor inflammation. Vasc. Cell 2012, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Jackaman, C.; Nelson, D.J. Are macrophages, myeloid derived suppressor cells and neutrophils mediators of local suppression in healthy and cancerous tissues in aging hosts? Exp. Gerontol. 2014, 54, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Gupta, K.; Pisarev, V.; Kinarsky, L.; Sherman, S.; Kang, L.; Herber, D.L.; Schneck, J.; Gabrilovich, D.I. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat. Med. 2007, 13, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S.; Sinha, P.; Beury, D.W.; Clements, V.K. Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin. Cancer Biol. 2012, 22, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Porta, C.; Morlacchi, S.; Banfi, S.; Strauss, L.; Rimoldi, M.; Totaro, M.G.; Riboldi, E. Origin and functions of tumor-associated myeloid cells (TAMCS). Cancer Microenviron. 2012, 5, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Poschke, I.; Wennerberg, E.; Pico de Coana, Y.; Egyhazi Brage, S.; Schultz, I.; Hansson, J.; Masucci, G.; Lundqvist, A.; Kiessling, R. Melanoma-educated CD14+ cells acquire a myeloid-derived suppressor cell phenotype through COX-2-dependent mechanisms. Cancer Res. 2013, 73, 3877–3887. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Gabrilovich, D.I. Hypoxia-inducible factors in regulation of immune responses in tumour microenvironment. Immunology 2014, 143, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Zou, L.; Rodriguez, P.; Zhu, G.; Wei, S.; Mottram, P.; Brumlik, M.; Cheng, P.; Curiel, T.; Myers, L.; et al. B7-H4 expression identifies a novel suppressive macrophage population in human ovarian carcinoma. J. Exp. Med. 2006, 203, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.M.; Vieweg, J.; Kusmartsev, S. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011, 71, 6400–6409. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, M.R.; Garlanda, C.; Jaillon, S.; Marone, G.; Mantovani, A. Tumor associated macrophages and neutrophils in tumor progression. J. Cell. Physiol. 2013, 228, 1404–1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, Q.; Qin, L.; Zhao, S.; Wang, J.; Chen, X. Transition of tumor-associated macrophages from MHC class II(hi) to MHC class II(low) mediates tumor progression in mice. BMC Immunol. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Day, Y.J.; Sag, D.; Linden, J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Chintala, N.K.; Vadrevu, S.K.; Patel, J.; Karbowniczek, M.; Markiewski, M.M. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J. Immunol. 2015, 194, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Ugel, S.; Peranzoni, E.; Desantis, G.; Chioda, M.; Walter, S.; Weinschenk, T.; Ochando, J.C.; Cabrelle, A.; Mandruzzato, S.; Bronte, V. Immune tolerance to tumor antigens occurs in a specialized environment of the spleen. Cell. Rep. 2012, 2, 628–639. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Lee, J.; Kim, J.; Jo, S.; Kim, Y.J.; Baek, J.E.; Kwon, E.S.; Lee, K.P.; Yang, S.; Kwon, K.S.; et al. Pancreatic adenocarcinoma up-regulated factor (PAUF) enhances the accumulation and functional activity of myeloid-derived suppressor cells (MDSCs) in pancreatic cancer. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Pucci, F.; Pittet, M.J. Molecular pathways: Tumor-derived microvesicles and their interactions with immune cells in vivo. Clin. Cancer Res. 2013, 19, 2598–2604. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Poschke, I.; Kiessling, R. Tumour-induced immune suppression: Role of inflammatory mediators released by myelomonocytic cells. J. Intern. Med. 2014, 276, 154–170. [Google Scholar] [CrossRef] [PubMed]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; et al. A model of human tumor dormancy: An angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.Y.; Li, J.F.; Gnatovskiy, L.; Deng, Y.; Zhu, L.; Grzesik, D.A.; Qian, H.; Xue, X.N.; Pollard, J.W. Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res. 2006, 66, 11238–11246. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, I.; Jose Carnicer, M.; Catasus, L.; Canet, B.; D’Angelo, E.; Zannoni, G.F.; Prat, J. Myometrial invasion and lymph node metastasis in endometrioid carcinomas: Tumor-associated macrophages, microvessel density, and HIF1A have a crucial role. Am. J. Surg. Pathol. 2010, 34, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Wu, X.; Zhong, C.; Yu, L.; Liang, X.H.; Yao, J.; Blanchard, D.; Bais, C.; Peale, F.V.; van Bruggen, N.; et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 2007, 450, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Varney, M.L.; Olsen, K.J.; Mosley, R.L.; Singh, R.K. Paracrine regulation of vascular endothelial growth factor—A expression during macrophage-melanoma cell interaction: Role of monocyte chemotactic protein-1 and macrophage colony-stimulating factor. J. Interferon Cytokine Res. 2005, 25, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Toge, H.; Inagaki, T.; Kojimoto, Y.; Shinka, T.; Hara, I. Angiogenesis in renal cell carcinoma: The role of tumor-associated macrophages. Int. J. Urol. 2009, 16, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Staudt, N.D.; Jo, M.; Hu, J.; Bristow, J.M.; Pizzo, D.P.; Gaultier, A.; VandenBerg, S.R.; Gonias, S.L. Myeloid cell receptor LRP1/CD91 regulates monocyte recruitment and angiogenesis in tumors. Cancer Res. 2013, 73, 3902–3912. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.R.; Corso, S.; Caione, L.; Cepero, V.; Conrotto, P.; Cignetti, A.; Piacibello, W.; Kumanogoh, A.; Kikutani, H.; Comoglio, P.M.; et al. Tumor angiogenesis and progression are enhanced by SEMA4D produced by tumor-associated macrophages. J. Exp. Med. 2008, 205, 1673–1685. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.; Raja, R.; Thorat, D.; Soundararajan, G.; Patil, T.V.; Kundu, G.C. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via α9β1 integrin. Oncogene 2014, 33, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Ojalvo, L.S.; Whittaker, C.A.; Condeelis, J.S.; Pollard, J.W. Gene expression analysis of macrophages that facilitate tumor invasion supports a role for Wnt-signaling in mediating their activity in primary mammary tumors. J. Immunol. 2010, 184, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Binsfeld, M.; Muller, J.; Lamour, V.; de Veirman, K.; de Raeve, H.; Bellahcene, A.; van Valckenborgh, E.; Baron, F.; Beguin, Y.; Caers, J.; et al. Granulocytic myeloid-derived suppressor cells promote angiogenesis in the context of multiple myeloma. Oncotarget 2016, 7, 37931–37943. [Google Scholar] [CrossRef] [PubMed]
- Venneri, M.A.; de Palma, M.; Ponzoni, M.; Pucci, F.; Scielzo, C.; Zonari, E.; Mazzieri, R.; Doglioni, C.; Naldini, L. Identification of proangiogenic TIE2-expressing monocytes (TEMs) in human peripheral blood and cancer. Blood 2007, 109, 5276–5285. [Google Scholar] [CrossRef] [PubMed]
- Lewis, C.E.; de Palma, M.; Naldini, L. TIE2-expressing monocytes and tumor angiogenesis: Regulation by hypoxia and angiopoietin-2. Cancer Res. 2007, 67, 8429–8432. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Toh, B.; Wang, X.; Keeble, J.; Sim, W.J.; Khoo, K.; Wong, W.C.; Kato, M.; Prevost-Blondel, A.; Thiery, J.P.; Abastado, J.P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011, 9, e1001162. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.L.; Ye, S.B.; OuYang, L.Y.; Zhang, H.; Chen, Y.S.; He, J.; Chen, Q.Y.; Qian, C.N.; Zhang, X.S.; Cui, J.; et al. Cox-2 promotes metastasis in nasopharyngeal carcinoma by mediating interactions between cancer cells and myeloid-derived suppressor cells. Oncoimmunology 2015, 4, e1044712. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Carmona, M.; Bourcy, M.; Lesage, J.; Leroi, N.; Syne, L.; Blacher, S.; Hubert, P.; Erpicum, C.; Foidart, J.M.; Delvenne, P.; et al. Soluble factors regulated by epithelial-mesenchymal transition mediate tumour angiogenesis and myeloid cell recruitment. J. Pathol. 2015, 236, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Nassar, D.; Blanpain, C. Cancer stem cells: Basic concepts and therapeutic implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Rong, Y.; Teng, Y.; Zhuang, X.; Samykutty, A.; Mu, J.; Zhang, L.; Cao, P.; Yan, J.; Miller, D.; et al. Exosomes miR-126a released from MDSC induced by DOX treatment promotes lung metastasis. Oncogene 2016. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Wilson, J.; Kulbe, H.; Li, N.F.; Leinster, D.A.; Charles, K.; Klemm, F.; Pukrop, T.; Binder, C.; Balkwill, F.R. Macrophages induce invasiveness of epithelial cancer cells via NF-κB and JNK. J. Immunol. 2005, 175, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Pukrop, T.; Klemm, F.; Hagemann, T.; Gradl, D.; Schulz, M.; Siemes, S.; Trumper, L.; Binder, C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. USA 2006, 103, 5454–5459. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lin, C.J.; Chen, K.H.; Wu, J.C.; Huang, S.H.; Wang, S.M. Macrophage activation increases the invasive properties of hepatoma cells by destabilization of the adherens junction. FEBS Lett. 2006, 580, 3042–3050. [Google Scholar] [CrossRef] [PubMed]
- Tjiu, J.W.; Chen, J.S.; Shun, C.T.; Lin, S.J.; Liao, Y.H.; Chu, C.Y.; Tsai, T.F.; Chiu, H.C.; Dai, Y.S.; Inoue, H.; et al. Tumor-associated macrophage-induced invasion and angiogenesis of human basal cell carcinoma cells by cyclooxygenase-2 induction. J. Investig. Dermatol. 2009, 129, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.W.; Ager, E.I.; Christophi, C. Bimodal role of Kupffer cells during colorectal cancer liver metastasis. Cancer Biol. Ther. 2013, 14, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Riboldi, E.; Porta, C.; Morlacchi, S.; Viola, A.; Mantovani, A.; Sica, A. Hypoxia-mediated regulation of macrophage functions in pathophysiology. Int. Immunol. 2013, 25, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Odegaard, J.I.; Chawla, A. Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Hardie, D.G. Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 2013, 493, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Yoza, B.K.; El Gazzar, M.; Vachharajani, V.T.; McCall, C.E. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J. Biol. Chem. 2011, 286, 9856–9864. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Devi, K.P.; Rajavel, T.; Russo, G.L.; Daglia, M.; Nabavi, S.F.; Nabavi, S.M. Molecular targets of omega-3 fatty acids for cancer therapy. Anticancer Agents Med. Chem. 2015, 15, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Azrad, M.; Turgeon, C.; Demark-Wahnefried, W. Current evidence linking polyunsaturated fatty acids with cancer risk and progression. Front. Oncol. 2013, 3, 224. [Google Scholar] [CrossRef] [PubMed]
- Antal, O.; Peter, M.; Hackler, L., Jr.; Man, I.; Szebeni, G.; Ayaydin, F.; Hideghety, K.; Vigh, L.; Kitajka, K.; Balogh, G.; et al. Lipidomic analysis reveals a radiosensitizing role of γ-linolenic acid in glioma cells. Biochim. Biophys. Acta 2015, 1851, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Yang, Q.; Shi, M.; Zhong, L.; Wu, C.; Meng, T.; Yin, H.; Zhou, J. Polyunsaturated fatty acids promote the expansion of myeloid-derived suppressor cells by activating the JAK/STAT3 pathway. Eur. J. Immunol. 2013, 43, 2943–2955. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Niino, D.; Saito, Y.; Ohnishi, K.; Horlad, H.; Ohshima, K.; Takeya, M. Clinical significance of CD163+ tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. 2013, 104, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Cairo, G.; Recalcati, S.; Mantovani, A.; Locati, M. Iron trafficking and metabolism in macrophages: Contribution to the polarized phenotype. Trends Immunol. 2011, 32, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of metabolism by macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed]
- OuYang, L.Y.; Wu, X.J.; Ye, S.B.; Zhang, R.X.; Li, Z.L.; Liao, W.; Pan, Z.Z.; Zheng, L.M.; Zhang, X.S.; Wang, Z.; et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J. Transl. Med. 2015, 13, 47. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic reprogramming of immune cells in cancer progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.; Ghossein, R.A.; Ricarte-Filho, J.C.; Knauf, J.A.; Fagin, J.A. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr. Relat. Cancer 2008, 15, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tumor-associated macrophages and survival in classic Hodgkin’s lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Fukushi, J.; Yamamoto, S.; Matsumoto, Y.; Setsu, N.; Oda, Y.; Yamada, H.; Okada, S.; Watari, K.; Ono, M.; et al. Macrophage infiltration predicts a poor prognosis for human ewing sarcoma. Am. J. Pathol. 2011, 179, 1157–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yao, G.; Zhang, Y.; Gao, J.; Yang, B.; Rao, Z. M2-polarized tumor-associated macrophages are associated with poor prognoses resulting from accelerated lymphangiogenesis in lung adenocarcinoma. Clinics 2011, 66, 1879–1886. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.C.; Liao, H.; Lin, S.X.; Xia, Y.; Wang, X.X.; Gao, Y.; Lin, Z.X.; Lu, J.B.; Huang, H.Q. High expression of tumor-infiltrating macrophages correlates with poor prognosis in patients with diffuse large B-cell lymphoma. Med. Oncol. 2012, 29, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.M.; Tan, Y.C.; Beretta, O.; Jiang, D.; Yeap, W.H.; Tai, J.J.; Wong, W.C.; Yang, H.; Schwarz, H.; Lim, K.H.; et al. Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur. J. Immunol. 2012, 42, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M.; Manolova, I.; Ananiev, J. The density of macrophages in colorectal cancer is inversely correlated to TGF-beta1 expression and patients’ survival. Virchows Arch. 2012, 461, S178. [Google Scholar]
- Kang, J.C.; Chen, J.S.; Lee, C.H.; Chang, J.J.; Shieh, Y.S. Intratumoral macrophage counts correlate with tumor progression in colorectal cancer. J. Surg. Oncol. 2010, 102, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Gabitass, R.F.; Annels, N.E.; Stocken, D.D.; Pandha, H.A.; Middleton, G.W. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol. Immunother. 2011, 60, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Weide, B.; Martens, A.; Zelba, H.; Stutz, C.; Derhovanessian, E.; di Giacomo, A.M.; Maio, M.; Sucker, A.; Schilling, B.; Schadendorf, D.; et al. Myeloid-derived suppressor cells predict survival of patients with advanced melanoma: Comparison with regulatory T cells and NY-ESO-1- or melan-A-specific T cells. Clin. Cancer Res. 2014, 20, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Gazzaniga, S.; Bravo, A.I.; Guglielmotti, A.; van Rooijen, N.; Maschi, F.; Vecchi, A.; Mantovani, A.; Mordoh, J.; Wainstok, R. Targeting tumor-associated macrophages and inhibition of MCP-1 reduce angiogenesis and tumor growth in a human melanoma xenograft. J. Investig. Dermatol. 2007, 127, 2031–2041. [Google Scholar] [CrossRef] [PubMed]
- Zollo, M.; di Dato, V.; Spano, D.; de Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Kapoor, V.; Buchlis, G.; Cheng, G.; Sun, J.; Wang, L.C.; Singhal, S.; Snyder, L.A.; Albelda, S.M. Monocyte chemoattractant protein-1 blockade inhibits lung cancer tumor growth by altering macrophage phenotype and activating CD8+ cells. Am. J. Respir. Cell Mol. Biol. 2011, 44, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- Ryder, M.; Gild, M.; Hohl, T.M.; Pamer, E.; Knauf, J.; Ghossein, R.; Joyce, J.A.; Fagin, J.A. Genetic and pharmacological targeting of CSF-1/CSF-1R inhibits tumor-associated macrophages and impairs BRAF-induced thyroid cancer progression. PLoS ONE 2013, 8, e54302. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 regulates tumor-promoting “emergency″ granulo-monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilling, B.; Sucker, A.; Griewank, K.; Zhao, F.; Weide, B.; Gorgens, A.; Giebel, B.; Schadendorf, D.; Paschen, A. Vemurafenib reverses immunosuppression by myeloid derived suppressor cells. Int. J. Cancer 2013, 133, 1653–1663. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Accart, N.; Kintz, J.; Cochin, S.; Reymann, C.; Le Pogam, F.; Marchand, J.B.; Menguy, T.; Slos, P.; Rooke, R.; et al. Therapeutic effects of anti-CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor-associated macrophages and osteoclasts. PLoS ONE 2013, 8, e73310. [Google Scholar] [CrossRef] [PubMed]
- Lohela, M.; Casbon, A.J.; Olow, A.; Bonham, L.; Branstetter, D.; Weng, N.; Smith, J.; Werb, Z. Intravital imaging reveals distinct responses of depleting dynamic tumor-associated macrophage and dendritic cell subpopulations. Proc. Natl. Acad. Sci. USA 2014, 111, E5086–E5095. [Google Scholar] [CrossRef] [PubMed]
- Rietkotter, E.; Menck, K.; Bleckmann, A.; Farhat, K.; Schaffrinski, M.; Schulz, M.; Hanisch, U.K.; Binder, C.; Pukrop, T. Zoledronic acid inhibits macrophage/microglia-assisted breast cancer cell invasion. Oncotarget 2013, 4, 1449–1460. [Google Scholar] [CrossRef] [PubMed]
- Coscia, M.; Quaglino, E.; Iezzi, M.; Curcio, C.; Pantaleoni, F.; Riganti, C.; Holen, I.; Monkkonen, H.; Boccadoro, M.; Forni, G.; et al. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J. Cell. Mol. Med. 2010, 14, 2803–2815. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wei, C.; Liu, Y.; Qiu, Y.; Liu, C.; Guo, F. Selective ablation of tumor-associated macrophages suppresses metastasis and angiogenesis. Cancer Sci. 2013, 104, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Luo, Y.; Sun, C.; Liu, Y.; Kuo, P.; Varga, J.; Xiang, R.; Reisfeld, R.; Janda, K.D.; Edgington, T.S.; et al. Targeting cell-impermeable prodrug activation to tumor microenvironment eradicates multiple drug-resistant neoplasms. Cancer Res. 2006, 66, 970–980. [Google Scholar] [CrossRef] [PubMed]
- Guth, A.M.; Hafeman, S.D.; Elmslie, R.E.; Dow, S.W. Liposomal clodronate treatment for tumour macrophage depletion in dogs with soft-tissue sarcoma. Vet. Comp. Oncol. 2013, 11, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Germano, G.; Belgiovine, C.; D'Incalci, M.; Mantovani, A. Trabectedin: A drug from the sea that strikes tumor-associated macrophages. Oncoimmunology 2013, 2, e24614. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Kapoor, V.; Jassar, A.S.; Kaiser, L.R.; Albelda, S.M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 2005, 11, 6713–6721. [Google Scholar] [CrossRef] [PubMed]
- Le, H.K.; Graham, L.; Cha, E.; Morales, J.K.; Manjili, M.H.; Bear, H.D. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int. Immunopharmacol. 2009, 9, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Sasso, M.S.; Lollo, G.; Pitorre, M.; Solito, S.; Pinton, L.; Valpione, S.; Bastiat, G.; Mandruzzato, S.; Bronte, V.; Marigo, I.; et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials 2016, 96, 47–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the NLRP3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, Y.; Zhang, Y.; Shang, Y.; Gao, Q. MDSC-decreasing chemotherapy increases the efficacy of cytokine-induced killer cell immunotherapy in metastatic renal cell carcinoma and pancreatic cancer. Oncotarget 2016, 7, 4760–4769. [Google Scholar] [PubMed]
- Huang, X.; Cui, S.; Shu, Y. Cisplatin selectively downregulated the frequency and immunoinhibitory function of myeloid-derived suppressor cells in a murine B16 melanoma model. Immunol. Res. 2016, 64, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Espagnolle, N.; Barron, P.; Mandron, M.; Blanc, I.; Bonnin, J.; Agnel, M.; Kerbelec, E.; Herault, J.P.; Savi, P.; Bono, F.; et al. Specific inhibition of the VEGFR-3 tyrosine kinase by SAR131675 reduces peripheral and tumor associated immunosuppressive myeloid cells. Cancers 2014, 6, 472–490. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Wei, X.; Luo, M.; Yu, J.; Tong, A.; Ma, X.; Ye, T.; Deng, H.; Sang, Y.; Liang, X.; et al. Inhibition of a20 expression in tumor microenvironment exerts anti-tumor effect through inducing myeloid-derived suppressor cells apoptosis. Sci. Rep. 2015, 5, 16437. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Kolomeyevskaya, N.; Singel, K.L.; Grimm, M.J.; Moysich, K.B.; Daudi, S.; Grzankowski, K.S.; Lele, S.; Ylagan, L.; Webster, G.A.; et al. Targeting myeloid cells in the tumor microenvironment enhances vaccine efficacy in murine epithelial ovarian cancer. Oncotarget 2015, 6, 11310–11326. [Google Scholar] [CrossRef] [PubMed]
- Stiff, A.; Trikha, P.; Wesolowski, R.; Kendra, K.; Hsu, V.; Uppati, S.; McMichael, E.; Duggan, M.; Campbell, A.; Keller, K.; et al. Myeloid-derived suppressor cells express bruton’s tyrosine kinase and can be depleted in tumor-bearing hosts by ibrutinib treatment. Cancer Res. 2016, 76, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.S.; Zea, A.H.; Rini, B.I.; Ireland, J.L.; Elson, P.; Cohen, P.; Golshayan, A.; Rayman, P.A.; Wood, L.; Garcia, J.; et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin. Cancer Res. 2009, 15, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; Ma, G.; Gildener-Leapman, N.; Eisenstein, S.; Coakley, B.A.; Ozao, J.; Mandeli, J.; Divino, C.; Schwartz, M.; Sung, M.; et al. Myeloid-derived suppressor cells as an immune parameter in patients with concurrent sunitinib and stereotactic body radiotherapy. Clin. Cancer Res. 2015, 21, 4073–4085. [Google Scholar] [CrossRef] [PubMed]
- Draghiciu, O.; Boerma, A.; Hoogeboom, B.N.; Nijman, H.W.; Daemen, T. A rationally designed combined treatment with an alphavirus-based cancer vaccine, sunitinib and low-dose tumor irradiation completely blocks tumor development. Oncoimmunology 2015, 4, e1029699. [Google Scholar] [CrossRef] [PubMed]
- Draghiciu, O.; Nijman, H.W.; Hoogeboom, B.N.; Meijerhof, T.; Daemen, T. Sunitinib depletes myeloid-derived suppressor cells and synergizes with a cancer vaccine to enhance antigen-specific immune responses and tumor eradication. Oncoimmunology 2015, 4, e989764. [Google Scholar] [CrossRef] [PubMed]
- Medina-Echeverz, J.; Aranda, F.; Berraondo, P. Myeloid-derived cells are key targets of tumor immunotherapy. Oncoimmunology 2014, 3, e28398. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.K.; Egilmez, N.K.; Suttles, J.; Stout, R.D. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J. Immunol. 2007, 178, 1357–1362. [Google Scholar] [CrossRef] [PubMed]
- U'Ren, L.; Guth, A.; Kamstock, D.; Dow, S. Type I interferons inhibit the generation of tumor-associated macrophages. Cancer Immunol. Immunother. 2010, 59, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Saccani, A.; Schioppa, T.; Porta, C.; Biswas, S.K.; Nebuloni, M.; Vago, L.; Bottazzi, B.; Colombo, M.P.; Mantovani, A.; Sica, A. P50 nuclear factor-κB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006, 66, 11432–11440. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Yaddanapudi, K.; Putty, K.; Rendon, B.E.; Lamont, G.J.; Faughn, J.D.; Satoskar, A.; Lasnik, A.; Eaton, J.W.; Mitchell, R.A. Control of tumor-associated macrophage alternative activation by macrophage migration inhibitory factor. J Immunol 2013, 190, 2984–2993. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Lu, Y.; Xu, Y.; Xu, L.; Zheng, W.; Wu, Y.; Li, L.; Shen, P. Estrogen represses hepatocellular carcinoma (HCC) growth via inhibiting alternative activation of tumor-associated macrophages (TAMs). J. Biol. Chem. 2012, 287, 40140–40149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tian, W.; Cai, X.; Wang, X.; Dang, W.; Tang, H.; Cao, H.; Wang, L.; Chen, T. Hydrazinocurcumin encapsuled nanoparticles “re-educate” tumor-associated macrophages and exhibit anti-tumor effects on breast cancer following STAT3 suppression. PLoS ONE 2013, 8, e65896. [Google Scholar] [CrossRef] [PubMed]
- Hackler, L., Jr.; Ozsvari, B.; Gyuris, M.; Sipos, P.; Fabian, G.; Molnar, E.; Marton, A.; Farago, N.; Mihaly, J.; Nagy, L.I.; et al. The curcumin analog C-150, influencing NF-κB, UPR and Akt/Notch pathways has potent anticancer activity in vitro and in vivo. PLoS ONE 2016, 11, e0149832. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, R.; Raina, D.; Meyer, C.; Kufe, D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1) → signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res. 2008, 68, 2920–2926. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, S.; Youn, J.I.; Weber, H.; Iclozan, C.; Lu, L.; Cotter, M.J.; Meyer, C.; Becerra, C.R.; Fishman, M.; Antonia, S.; et al. Anti-inflammatory triterpenoid blocks immune suppressive function of mdscs and improves immune response in cancer. Clin. Cancer Res. 2010, 16, 1812–1823. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Jassar, A.; Mishalian, I.; Wang, L.C.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Levy, L.; Albelda, S.M. Using macrophage activation to augment immunotherapy of established tumours. Br. J. Cancer 2013, 108, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Albeituni, S.H.; Ding, C.; Liu, M.; Hu, X.; Luo, F.; Kloecker, G.; Bousamra, M., 2nd; Zhang, H.G.; Yan, J. Yeast-derived particulate β-glucan treatment subverts the suppression of myeloid-derived suppressor cells (MDSC) by inducing polymorphonuclear MDSC apoptosis and monocytic MDSC differentiation to apc in cancer. J. Immunol. 2016, 196, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Ma, J.; Ma, K.; Guo, H.; Baidoo, S.E.; Zhang, Y.; Yan, J.; Lu, L.; Xu, H.; Wang, S. Beta-glucan enhances antitumor immune responses by regulating differentiation and function of monocytic myeloid-derived suppressor cells. Eur. J. Immunol. 2013, 43, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.A.; Lie, J.D. Phosphodiesterase-5 (PDE5) inhibitors in the management of erectile dysfunction. Pharm. Ther. 2013, 38, 407–419. [Google Scholar]
- Giannetta, E.; Feola, T.; Gianfrilli, D.; Pofi, R.; Dall’Armi, V.; Badagliacca, R.; Barbagallo, F.; Lenzi, A.; Isidori, A.M. Is chronic inhibition of phosphodiesterase type 5 cardioprotective and safe? A meta-analysis of randomized controlled trials. BMC Med. 2014, 12, 185. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Meckel, K.; Kelso, M.; Noonan, K.; Califano, J.; Koch, W.; Dolcetti, L.; Bronte, V.; Borrello, I. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 2006, 203, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Huang, X.; Lynn, K.D.; Thorpe, P.E. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol. Res. 2013, 1, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Pico de Coana, Y.; Poschke, I.; Gentilcore, G.; Mao, Y.; Nystrom, M.; Hansson, J.; Masucci, G.V.; Kiessling, R. Ipilimumab treatment results in an early decrease in the frequency of circulating granulocytic myeloid-derived suppressor cells as well as their arginase1 production. Cancer Immunol. Res. 2013, 1, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Cagnon, L.; Costa-Nunes, C.M.; Baumgaertner, P.; Montandon, N.; Leyvraz, L.; Michielin, O.; Romano, E.; Speiser, D.E. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol. Immunother. 2014, 63, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Martens, A.; Wistuba-Hamprecht, K.; Geukes Foppen, M.; Yuan, J.; Postow, M.A.; Wong, P.; Romano, E.; Khammari, A.; Dreno, B.; Capone, M.; et al. Baseline peripheral blood biomarkers associated with clinical outcome of advanced melanoma patients treated with ipilimumab. Clin. Cancer Res. 2016, 22, 2908–2918. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Freeman, G.J.; Coukos, G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res. 2013, 73, 6900–6912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Soto, A.; de las Casas-Engel, M.; Bragado, R.; Medina-Echeverz, J.; Aragoneses-Fenoll, L.; Martin-Gayo, E.; van Rooijen, N.; Berraondo, P.; Toribio, M.L.; Moro, M.A.; et al. Intravenous immunoglobulin promotes antitumor responses by modulating macrophage polarization. J. Immunol. 2014, 193, 5181–5189. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Liang, P.; Guo, G.; Huang, Z.; Niu, Y.; Dong, L.; Wang, C.; Zhang, J. Re-polarizing myeloid-derived suppressor cells (MDSCs) with cationic polymers for cancer immunotherapy. Sci. Rep. 2016, 6, 24506. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Oliver, L.; Alvarez, R.; Fernandez, L.E.; Lee, K.P.; Mesa, C. Adjuvants and myeloid-derived suppressor cells: Enemies or allies in therapeutic cancer vaccination. Hum. Vaccines Immunother. 2014, 10, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Amoozgar, Z.; Goldberg, M.S. Targeting myeloid cells using nanoparticles to improve cancer immunotherapy. Adv. Drug Deliv. Rev. 2015, 91, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Shirota, Y.; Shirota, H.; Klinman, D.M. Intratumoral injection of CpG oligonucleotides induces the differentiation and reduces the immunosuppressive activity of myeloid-derived suppressor cells. J. Immunol. 2012, 188, 1592–1599. [Google Scholar] [CrossRef] [PubMed]
- Seya, T.; Shime, H.; Matsumoto, M. Functional alteration of tumor-infiltrating myeloid cells in rna adjuvant therapy. Anticancer Res. 2015, 35, 4385–4392. [Google Scholar] [PubMed]
- Chuang, C.M.; Monie, A.; Hung, C.F.; Wu, T.C. Treatment with imiquimod enhances antitumor immunity induced by therapeutic HPV DNA vaccination. J. Biomed. Sci. 2010, 17, 32. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Armstrong, A.J. Tasquinimod in the treatment of castrate-resistant prostate cancer—Current status and future prospects. Ther. Adv. Urol. 2016, 8, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Sundstedt, A.; Ciesielski, M.; Miles, K.M.; Celander, M.; Adelaiye, R.; Orillion, A.; Ciamporcero, E.; Ramakrishnan, S.; Ellis, L.; et al. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol. Res. 2015, 3, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Larghi, P.; Porta, C.; Riboldi, E.; Totaro, M.G.; Carraro, L.; Orabona, C.; Sica, A. The p50 subunit of NF-κB orchestrates dendritic cell lifespan and activation of adaptive immunity. PLoS ONE 2012, 7, e45279. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Ruiz, J.R.; Baird, J.R.; Tesone, A.J.; Rutkowski, M.R.; Scarlett, U.K.; Camposeco-Jacobs, A.L.; Anadon-Arnillas, J.; Harwood, N.M.; Korc, M.; Fiering, S.N.; et al. Reprogramming tumor-associated dendritic cells in vivo using miRNA mimetics triggers protective immunity against ovarian cancer. Cancer Res. 2012, 72, 1683–1693. [Google Scholar] [CrossRef] [PubMed]
- Draghiciu, O.; Lubbers, J.; Nijman, H.W.; Daemen, T. Myeloid derived suppressor cells—An overview of combat strategies to increase immunotherapy efficacy. Oncoimmunology 2015, 4, e954829. [Google Scholar] [CrossRef] [PubMed]
- Rivera, L.B.; Meyronet, D.; Hervieu, V.; Frederick, M.J.; Bergsland, E.; Bergers, G. Intratumoral myeloid cells regulate responsiveness and resistance to antiangiogenic therapy. Cell Rep. 2015, 11, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Curtis, V.F.; Wang, H.; Yang, P.; McLendon, R.E.; Li, X.; Zhou, Q.Y.; Wang, X.F. A PK2/Bv8/PROK2 antagonist suppresses tumorigenic processes by inhibiting angiogenesis in glioma and blocking myeloid cell infiltration in pancreatic cancer. PLoS ONE 2013, 8, e54916. [Google Scholar] [CrossRef] [PubMed]
- Paris, F.; Fuks, Z.; Kang, A.; Capodieci, P.; Juan, G.; Ehleiter, D.; Haimovitz-Friedman, A.; Cordon-Cardo, C.; Kolesnick, R. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science 2001, 293, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Keefe, D.M.; Brealey, J.; Goland, G.J.; Cummins, A.G. Chemotherapy for cancer causes apoptosis that precedes hypoplasia in crypts of the small intestine in humans. Gut 2000, 47, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Benoit, M.; Desnues, B.; Mege, J.L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [PubMed]
- Mege, J.L.; Mehraj, V.; Capo, C. Macrophage polarization and bacterial infections. Curr. Opin. Infect. Dis. 2011, 24, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Immunosuppressive drugs: The first 50 years and a glance forward. Immunopharmacology 2000, 47, 63–83. [Google Scholar] [CrossRef]
- Kienle, G.S. Fever in cancer treatment: Coley’s therapy and epidemiologic observations. Glob. Adv. Health Med. 2012, 1, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Viaud, S.; Daillere, R.; Boneca, I.G.; Lepage, P.; Langella, P.; Chamaillard, M.; Pittet, M.J.; Ghiringhelli, F.; Trinchieri, G.; Goldszmid, R.; et al. Gut microbiome and anticancer immune response: Really hot sh*t! Cell Death Differ. 2015, 22, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, C.M.; Shih, J.H.; Astin, J.W.; Rewcastle, G.W.; Flanagan, J.U.; Crosier, P.S.; Shepherd, P.R. DMXAA (vadimezan, ASA404) is a multi-kinase inhibitor targeting VEGFR2 in particular. Clin. Sci. 2012, 122, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Raymond, E.; Dalgleish, A.; Damber, J.E.; Smith, M.; Pili, R. Mechanisms of action of tasquinimod on the tumour microenvironment. Cancer Chemother. Pharmacol. 2014, 73, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Masciulli, R.; Tritarelli, E.; Pustorino, R.; Mariani, G.; Martucci, R.; Barberi, T.; Camagna, A.; Valtieri, M.; Peschle, C. Transforming growth factor-β potentiates vitamin D3-induced terminal monocytic differentiation of human leukemic cell lines. J. Immunol. 1993, 150, 2418–2430. [Google Scholar] [PubMed]
- Young, M.R.; Ihm, J.; Lozano, Y.; Wright, M.A.; Prechel, M.M. Treating tumor-bearing mice with vitamin D3 diminishes tumor-induced myelopoiesis and associated immunosuppression, and reduces tumor metastasis and recurrence. Cancer Immunol. Immunother. 1995, 41, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Lathers, D.M.; Clark, J.I.; Achille, N.J.; Young, M.R. Phase 1B study to improve immune responses in head and neck cancer patients using escalating doses of 25-hydroxyvitamin D3. Cancer Immunol. Immunother. 2004, 53, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Velders, M.P.; Sotomayor, E.M.; Kast, W.M. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J. Immunol. 2001, 166, 5398–5406. [Google Scholar] [CrossRef] [PubMed]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-trans-retinoic acid improves differentiation of myeloid cells and immune response in cancer patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Park, C.S.; Lee, Y.R.; Im, S.A.; Song, S.; Lee, C.K. Resiquimod, a TLR7/8 agonist, promotes differentiation of myeloid-derived suppressor cells into macrophages and dendritic cells. Arch. Pharm. Res. 2014, 37, 1234–1240. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, Y.; Nakatsuji, M.; Seno, H.; Ishizu, S.; Akitake-Kawano, R.; Kanda, K.; Ueo, T.; Komekado, H.; Kawada, M.; Minami, M.; et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis 2011, 32, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Yoon, Y.N.; Son, D.I.; Seok, S.H. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS ONE 2013, 8, e63451. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cell function is reduced by withaferin A, a potent and abundant component of Withania somnifera root extract. Cancer Immunol. Immunother. 2013, 62, 1663–1673. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Geng, Z.; Dominguez, D.; Chen, S.; Fan, J.; Qin, L.; Long, A.; Zhang, Y.; Kuzel, T.M.; Zhang, B. Targeting ornithine decarboxylase by alpha-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J. Immunol. 2016, 196, 915–923. [Google Scholar] [CrossRef] [PubMed]


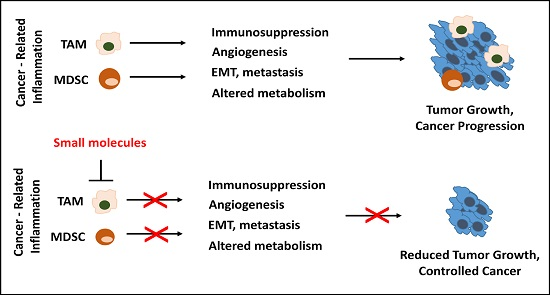{kind=link}
{kind=link}
| Compounds | Chemical Structures | In Vivo Effect | Mechanism of Action | References |
|---|---|---|---|---|
| Bindarit |  | Decreases the infiltration of TAMs and MDSCs | Inhibits the synthesis of C-C motif chemokine ligand 2 (CCL2) | [121,122] |
| GW2850 |  | Decreases the infiltration of TAMs and MDSCs | Selective receptor kinase CSF1R (CD115) inhibitor | [124,125,126] |
| PLX3397 (Pexidartinib) |  | Decreases the infiltration of TAMs and MDSCs | Selective receptor kinase CSF1R (CD115) inhibitor | [124,126] |
| Vemurafenib |  | Blocks the recruitment of both M-MDSCs and G-MDSCs | Selective B-Raf kinase inhibitor | [128] |
| Compounds | Chemical Structures | In Vivo Effect | Mechanism of Action | References |
|---|---|---|---|---|
| Zoledronic acid |  | Reduced the number of TAMs and reverted their polarization from M2 to M1 | Inhibits the active site of the enzyme farnesyl pyrophosphate (FPP) synthase in the mevalonate (Mev) pathway | [131,132] |
| Doxorubicin-based prodrug (LEG-3) |  | Depletes TAMs | LEG-3 is a legumain, an asparagynil endopeptidase activated prodrug. Doxorubicin is a DNA intercalator | [133,134] |
| Clodronate (encapsulated liposomes) |  | Depletes TAMs | Clodronate is converted to non-hydrolyzable ATP analogue intracellularly | [121,135] |
| Trabectedin (Yondelis®) |  | Induces apoptosis of mononuclear phagocytes (TAMs, monocytes) | Caspase-8 activation via TRAIL-Rs pathway | [136,137] |
| Gemcitabine |  | Reduces the expansion of Gr1+/CD11b+ splenic MDSCs | Nucleoside analog | [139] |
| 5-fluorouracil (5-FU) |  | Causes apoptosis and depletion of MDSCs | Pyrimidine analog | [141] |
| Cisplatin |  | Depleted 50% of tumor infiltrating Gr1+/CD11b+ MDSCs | Forms DNA adducts | [144] |
| SAR131675 |  | Reduces the number of splenic Gr1+/CD11b+ cells and F4/80high TAMs | VEGFR-3 inhibitor | [145] |
| Ibrutinib |  | Inhibits the recruitment of CD11b+/Gr1+ MDSCs in the tumor and spleen | Irreversible inhibitor of Bruton’s tyrosin kinase (BTK) and IL-2 inducible T-cell kinase (ITK) | [148] |
| Sunitinib |  | Reduces MDSCs in the blood, enhances IFN-γ+ Th1 response and reduces T-regs | Multi-targeted receptor tyrosine kinase inhibitor | [149,150,151,152] |
| Compounds | Chemical Structures | In Vivo Effect | Mechanism of Action | References |
|---|---|---|---|---|
| 4-iodo-6-phenylpyrimidine (4-IPP) |  | Reduces ArgI and elevates TNF-α expression in TAM, attenuates TAM and both splenic Gr1high Ly6G+ G-MDSC and Gr1dim, Ly6G− M-MDSCs mediated immunosuppression | Migration inhibitory factor (MIF) antagonist | [158] |
| Hydrazinocurcumin |  | Re-educates TAMs to be IL-12high, IL-10low and TGF-βlow | Suppresses STAT3 | [160] |
| Bardoxolone methyl (CDDO-Me) |  | Abrogates the immune suppressive effect of MDSCs | JAK1 and STAT3 inhibitor | [162,163,196] |
| 5,6 Dimethylxanthenone-4-acetic-acid (DMXAA, Vadimezan or ASA404) |  | Increases the influx of neutrophils and anti-tumour (M1) macrophages to the tumour, induces macrophage activation, augments the therapeutic effects of immunotherapy | ‘Stimulator of interferon gene’ (STING) agonist, multi-kinase inhibitor | [164,197] |
| Sildenafil (Viagra®) |  | Down-regulates ArgI and NOS2 enzymatic activity of tumor infiltrating MDSCs | Phosphodiesterase-5 (PDE-5) inhibitor | [169] |
| Imiquimod |  | Decreases tumor infiltrating MDSCs and activates antitumor NK 1.1+ cells and F4/80+ macrophages in combination with immunotherapy | TLR7 agonist | [177,181] |
| Tasquinimod |  | Inhibits the accumulation of Ly6C+ MDSCs and CD206+ M2-like TAMs | Orally active S100A9 inhibitor | [182,183,198] |
| IPI145 (Duvelisib) |  | Enhances the efficacy of VEGF/VEGFR blockade anti-angiostatic therapy by sorafenib. IPI145 decreases intra-tumoral TAM, Gr1+ monocytes and tumor-associated neutrophils | Phosphatidylinositol-3 kinase γ and δ (PI3Kγ and δ) inhibitor | [187] |
| PKRA7 |  | Inhibits the neovascularization of glioma and myeloid cell infiltration of pancreatic cancer | Prokineticin 2 (PK2 or Bv8) antagonist | [188] |
| Compounds | Chemical Structures | In Vivo Effect | Mechanism of Action | References |
|---|---|---|---|---|
| D3 vitamin (Cholecalciferol) |  | Induces monocytic differentiation, reduces tumor-induced myelopoiesis, reduces the number of CD34+ immunosuppressive cells | Calcitriol (vitamin D) receptor agonist | [199,200,201] |
| ATRA (Tretinoin) |  | Combined with GM-CSF differentiates immature myeloid Gr1+ cells, eliminates their inhibitory potential | Retinoic acid receptor agonist | [202,203] |
| Resiquimod |  | Differentiates MDSCs to F4/80+ macrophages | TLR7/8 agonist | [177,204] |
| Compounds | Chemical Structures | In Vivo Effect | Mechanism of Action | References |
|---|---|---|---|---|
| Celecoxib |  | Reverted TAM phenotype from M2 to M1 | Cyclooxygenase II (COX-2) inhibitor | [50,205] |
| Etodolac |  | Blocks M2 macrophage differentiation and suppresses metastasis formation | COX-2 inhibitor | [206] |
| Tyrphostin AG490 |  | Decreases the T-cell inhibitory function of melanoma patient-derived CD14+ cells | Inhibits STAT3 phosphorylation | [63] |
| Withaferin A |  | Reduces IL-10 production of MDSCs and the accumulation of G-MDSCs | Inhibits ROS production via inhibition of STAT3 phosphorylation | [207] |
| N-acetyl cysteine (NAC) |  | Restores both CD4+ and CD8+ T-cell proliferation and activation | Antioxidant and enters cells via ASC transporters, rapidly hydrolyzes to cysteine | [208] |
| α-Difluoromethylornithine (DFMO) |  | Decreases ArgI production in MDSCs | Ornithine-decarboxylase (ODC) inhibitor | [209] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szebeni, G.J.; Vizler, C.; Nagy, L.I.; Kitajka, K.; Puskas, L.G. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int. J. Mol. Sci. 2016, 17, 1958. https://doi.org/10.3390/ijms17111958
Szebeni GJ, Vizler C, Nagy LI, Kitajka K, Puskas LG. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. International Journal of Molecular Sciences. 2016; 17(11):1958. https://doi.org/10.3390/ijms17111958
Chicago/Turabian StyleSzebeni, Gabor J., Csaba Vizler, Lajos I. Nagy, Klara Kitajka, and Laszlo G. Puskas. 2016. "Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets" International Journal of Molecular Sciences 17, no. 11: 1958. https://doi.org/10.3390/ijms17111958
APA StyleSzebeni, G. J., Vizler, C., Nagy, L. I., Kitajka, K., & Puskas, L. G. (2016). Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. International Journal of Molecular Sciences, 17(11), 1958. https://doi.org/10.3390/ijms17111958