
The Impact of CXCR4 Blockade on the Survival of Rat Brain Cortical Neurons

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Effect of AMD3100 on Cell Viability in Brain Cortical Neurons at 7 DIV (Days in Vitro)
Cell Treatment
2.2. AMD3100 Altered Mitochondrial Membrane Potential by Hyperpolarization in Rat Brain Cortical Neurons
2.3. Effect of AMD3100 on Lactate Dehydrogenase (LDH) Release in Rat Brain Cortical Neurons
2.4. Effect of AMD3100 on Reactive Oxygen Species (ROS) Production in Brain Cortical Neurons at 7 DIV
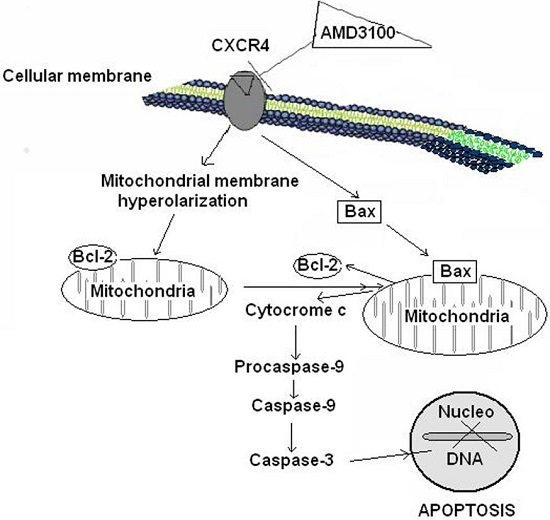
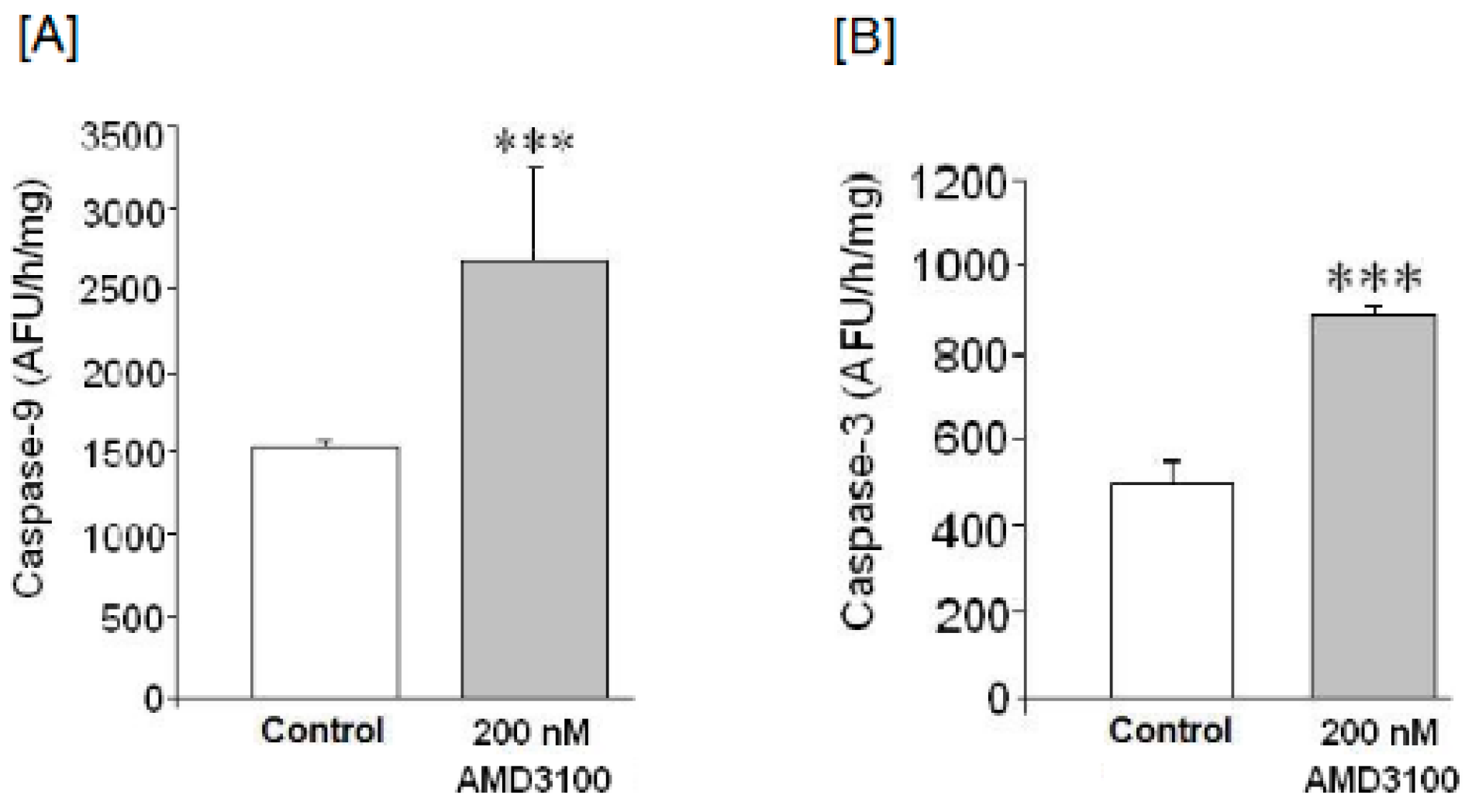
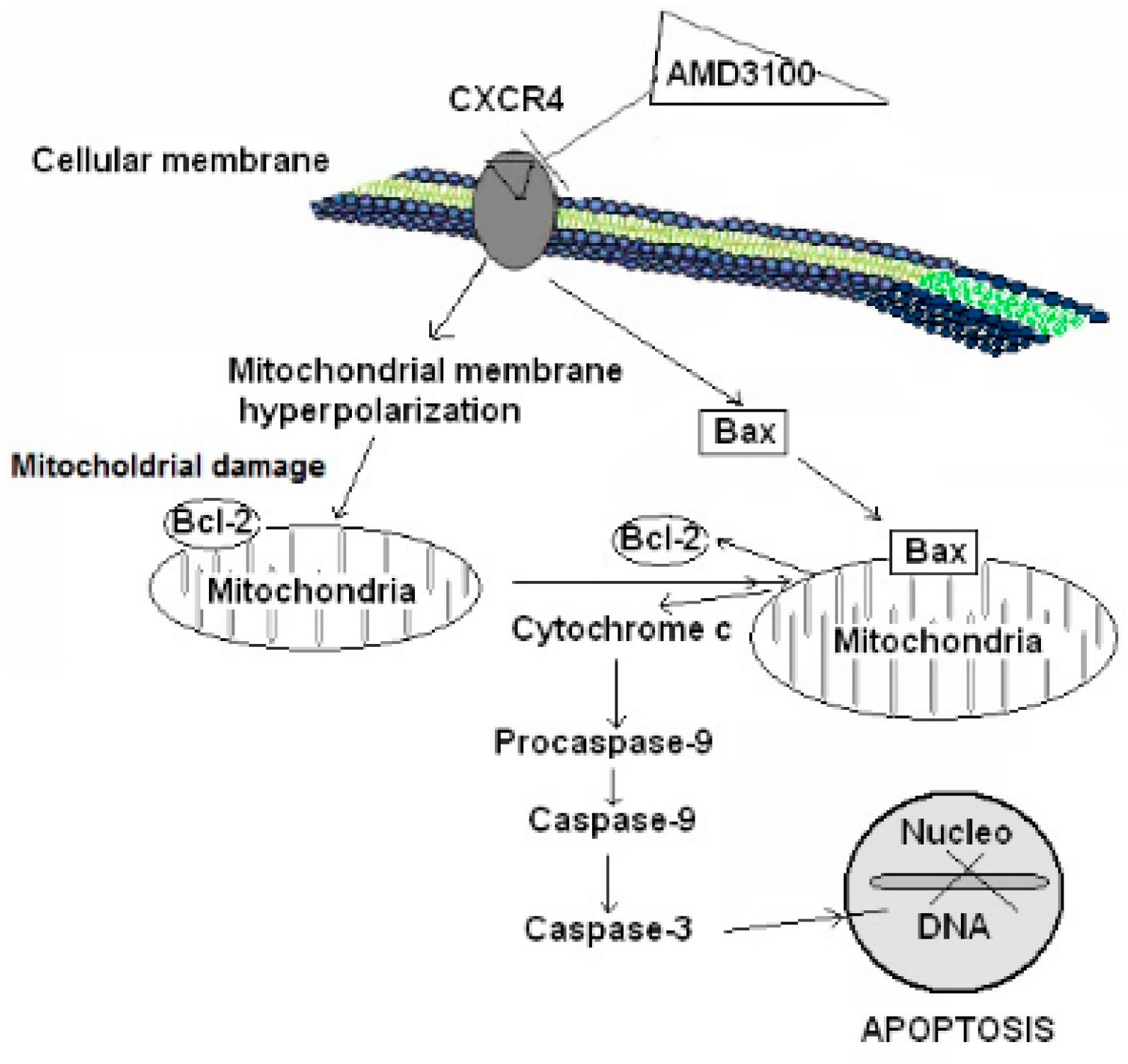
2.5. Effect of AMD3100 on Caspase-9 Activity in Rat Brain Cortical Neurons
2.6. Effect of AMD3100 on Caspase-3 Activity in Rat Cortical Neurons
2.7. Effect of AMD3100 on Cytochrome C Release in Rat Brain Cortical Neurons
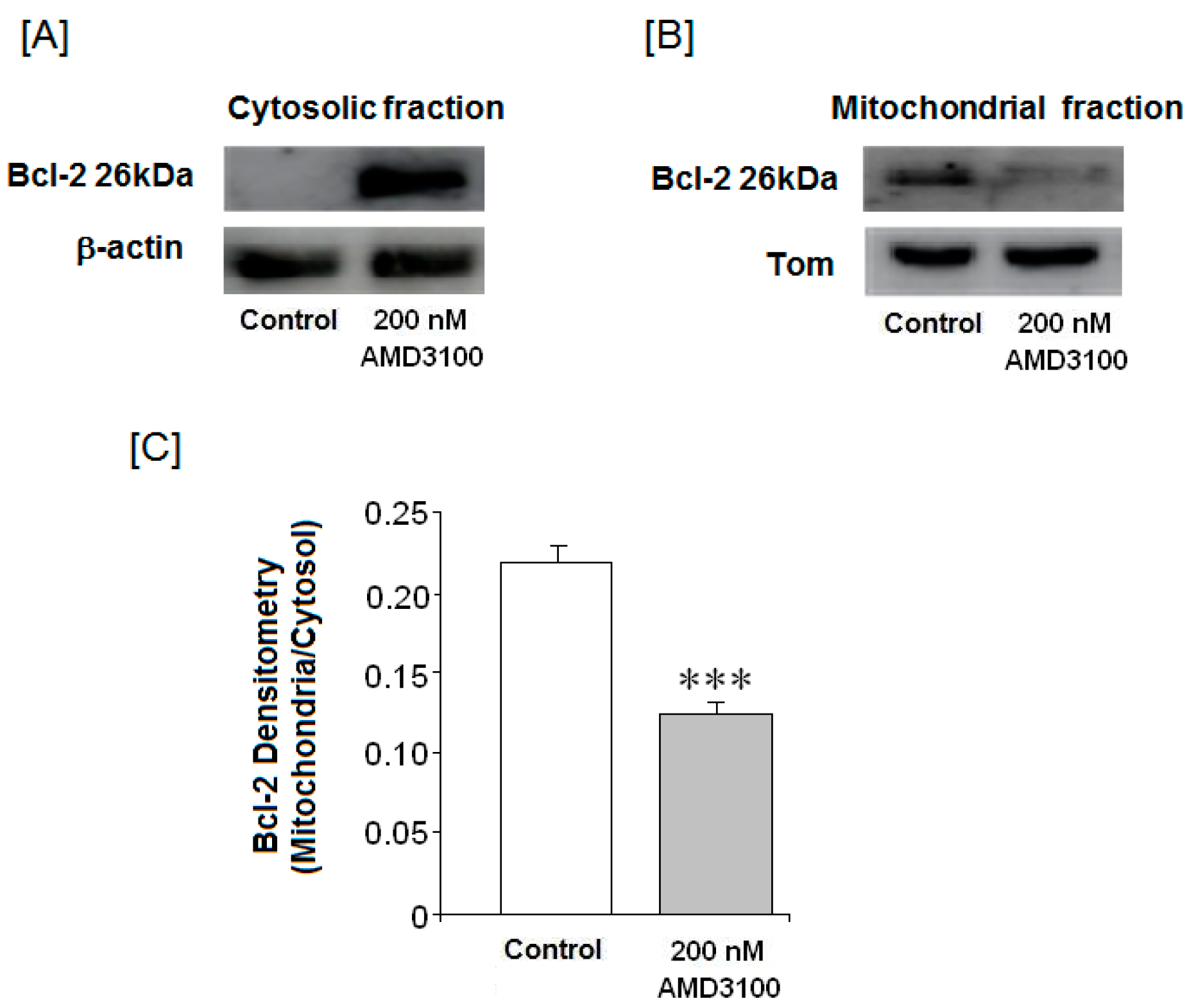
2.8. Effect of AMD3100 on Cytosolic and Mitochondrial Bax Protein Translocation in Rat Brain Cortical Neurons
2.9. Effect of AMD3100 on the Mitochondrial Bcl-2 Protein Levels in Rat Brain Cortical Neurons
3. Discussion
4. Material and Methods
4.1. Ethics Statement
4.2. Materials
4.3. Methods
4.3.1. Cell Isolation and Culture
4.3.2. Assessment of Cell Viability by XTT Assay
4.3.3. Measurement of ROS Formation
4.3.4. Cellular Death (LDH Assay)
4.3.5. Mitochondrial Membrane Potential Measurement (MMP)
4.3.6. Caspase-3 Activity Measurement
4.3.7. Caspase-9 Activity
4.4. Preparation of Cell Lysates (Subcellular Fractionation)
4.5. Protein Evaluation of Apoptotic Markers by Western Blot
4.6. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Donzella, G.A.; Schols, D.; Lin, S.W.; Esté, J.A.; Nagashima, K.A.; Maddon, P.J.; Allaway, G.P.; Sakmar, T.P.; Henson, G.; de Clercq, E.; et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat. Med. 1998, 4, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Liesveld, J.L.; Bechelli, J.; Rosell, K.; Lu, C.; Bridger, G.; Phillips, G., II; Abboud, C.N. Effects of AMD3100 on transmigration and survival of acute myelogenous leukemia cells. Leuk. Res. 2007, 31, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. CXCR4: A key receptor in the crosstalk between tumour cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.T.; Bartley, J.H.; Wimborne, H.J.; Walker, A.L.; Hess, D.C.; Hill, W.D.; Carroll, J.E. The neuroblast and angioblast chemotaxic factor SDF-1 (CXCL12) expression is briefly up regulated by reactive astrocytes in brain following neonatal hypoxic-ischemic injury. BMC Neurosci. 2005, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Merino, J.J.; Bellver-Landete, V.; Oset-Gasque, M.J.; Cubelos, B. CXCR4/CXCR7 molecular involvement in neuronal and neural progenitor migration: Focus in CNS repair. J. Cell. Physiol. 2015, 230, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.; Brandimarti, R.; Shimizu, S.; Nicolai, J.; Crowe, E.; Meucci, O. The chemokine CXCL12 promotes survival of postmitotic neurons by regulating Rb protein. Cell Death Differ. 2008, 15, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Lazarini, F.; Tham, T.N.; Casanova, P.; Arenzana-Seisdedos, F.; Dubois-Dalcq, M. Role of the α-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia 2003, 42, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Reaux-Le Goazigo, A.; van Steenwinckel, J.; Rostene, W.; Melik Parsadaniantz, S. Current status of chemokines in the adult CNS. Prog. Neurobiol. 2013, 104, 67–92. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.; Kitabi, P.; Boudin, H. The chemokine SDF-1α differntiatelly regulates axonal elongation and branching in hippocampal neurons. J. Cell Sci. 2004, 118, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Grove, E.A.; Miller, R.J. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 7090–7095. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Mu, J.W.; Wang, H.B.; Jolkkonen, J.; Liu, T.T.; Xiao, T.; Zhao, M.; Zhang, C.D.; Zhao, C.S. Increased protein expression levels of pCREB, BDNF and SDF-1/CXCR4 in the hippocampus may be associated with enhanced neurogenesis induced by environmental enrichment. Mol. Med. Rep. 2016, 14, 2231–2237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Qu, H.; Zhao, Y.; Xiao, T.; Zhao, M.; Li, Y.; Jolkkonen, J.; Cao, Y.; Zhao, C. CXCR4 antagonist AMD3100 reverses the neurogenesis and behavioral recovery promoted by forced limb-use in stroke rats. Restor. Neurol. Neurosci. 2015, 33, 809–821. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, Y.; Tang, Y.; Tang, G.; Yang, G.Y.; Wang, Y. CXCR4 antagonist AMD3100 protects blood-brain barrier integrity and reduces inflammatory response after focal ischemia in mice. Stroke 2013, 44, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich-Nikitin, I.; Ezra, A.; Barbiro, B.; Rabinovich-Toidman, P.; Solomon, B. Chronic administration of AMD3100 increases survival and alleviates pathology in SOD1G93A mice model of ALS. J. Neuroinflamm. 2016, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Menichella, D.M.; Abdelhak, B.; Ren, D.; Shum, A.; Frietag, C.; Miller, R.J. CXCR4 chemokine receptor signaling mediates pain in diabetic neuropathy. Mol. Pain 2014, 25, 10–42. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Wang, X.; Li, Z.; Kong, C.; Zhao, Y.; Qian, J.L.; Kan, Q.; Zhang, W.; Xu, J.T. Upregulation of Chemokine CXCL12 in the Dorsal Root Ganglia and Spinal Cord Contributes to the Development and Maintenance of Neuropathic Pain Following Spared Nerve Injury in Rats. Neurosci. Bull. 2016, 32, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wang, M.; Guo, Y.Y.; Sun, T.; Li, Y.J.; Yang, Q.; Zhang, K.; Liu, S.B.; Zhao, M.G.; Wu, Y.M. Systemic inflammation induces anxiety disorder through CXCL12/CXCR4 pathway. Brain Behav. Immun. 2016, 56, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Eng. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Apoptosis-based therapies. Nat. Rev. Drug Discov. 2002, 1, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008, 18, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, D.R.; Green, J.E. Apoptosis: Stabbed in the Bax. Nature 2008, 455, 195–200. [Google Scholar]
- Cory, S.; Adams, J.M. The Bcl-2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Saeed, S.; Quintin, J.; Kerstens, H.H.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Yang, G.; Hunter, Z.R.; Liu, X.; Xu, L.; Chen, J.; Tsakmaklis, N.; Hatjiharissi, E.; Kanan, S.; Davids, M.S.; et al. The BCL-2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4Wild-type and CXCR4WHIM mutated Waldenstrom macroglobulinaemia cells. Br. J. Haematol. 2015, 170, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Garcimartín, A.; José, J.; Merino, J.J.; González, M.P.; Sánchez-Reus, M.I.; Sánchez-Muniz, F.J.; Bastida, S.; Benedí, J. Organic silicon protects human neuroblastoma SH-SY5Y cells against hydrogen peroxide effects. BMC Complement. Altern. Med. 2014, 14, 384. [Google Scholar] [CrossRef] [PubMed]
- Garcimartin, A.; Merino, J.J.; Santos-López, J.A.; López-Oliva, M.E.; González, M.P.; Sánchez-Muñiz, F.J.; Benedí, J. Silicon as neuroprotector or neurotoxic in the human neuroblastoma SH-SY5Y cell line. Chemosphere 2015, 135, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.R.; Hu, L.S.; Li, G.Y. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar] [PubMed]
- Barbosa, F.J.; Queirós, O.; Moreira, R.; Carvalho, F.; Dinis-Oliveira, R.J. Comparative study of the neurotoxicological effects of tramadol and tapentadol in SH-SY5Y cells. Toxicology 2016, 359, 1–10. [Google Scholar]
- Zuang, H.; Li, Y.; Chi, Y. Role of p38 MAPK activation and mitochondrial cytochrome-c release in allicin-induced apoptosis in SK-N-SH cells. Anticancer Drugs 2014, 27, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Baoudouin, S.J.; Pujol, F.; Nicot, A.; Kitabgi, P.; Boudin, H. Dendrite-selective redistribution of thechemokine receptor CXCR4 following agonist stimulation. Mol. Cell. Neurosci. 2006, 33, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Fricker, S.P.; Anastassov, V.; Cox, J.; Darkes, M.C.; Grujic, O.; Idzan, S.R.; Labrecque, J.; Lau, G.; Mosi, R.M.; Nelson, K.L.; et al. Characterization of the molecular pharmacology of AMD3100: A specific antagonist of the G-protein coupled chemokine receptor, CXCR4 using CCRF-CEM T-cell line. Biochem. Pharmacol. 2006, 72, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Yi, X.; Qin, J.; Tian, M.; Jin, G. CXCL12 inhibits cortical neuron apoptosis by increasing the ratio of Bcl-2/Bax after traumatic brain injury. Int. J. Neurosci. 2014, 124, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Segal, M. Rat hippocampus neurons in culture: Responses to electrical and chemical stimuli. J. Neurophysiol. 1983, 50, 1249–1264. [Google Scholar] [PubMed]
- Figueroa, S.; Oset-Gasque, M.J.; Arce, C.; Martínez-Honduvilla, C.; González, M.P. Mitochondrial involvement in nitric oxide-induced cellular death in cortical neurons in culture. J. Neurosci. Res. 2006, 83, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Papassotiropoulos, A.; Ludwig, M.; Naib-Majani, W.; Rao, G.S. Induction of apoptosis and secondary necrosis in rat dorsal root ganglionar cell cultures by oxidized low density lipoprotein. Neurosci. Lett. 1996, 3, 33–36. [Google Scholar] [CrossRef]
- López, E.; Figueroa, S.; Oset-Gasque, M.J.; González, M.P. Apoptosis and necrosis: Two distinct events induced by cadmium in cortical neurons in culture. Br. J. Pharmacol. 2003, 138, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Tenneti, L.; Mc D’Emilia, D.; Troy, C.M.; Liption, S.A. Role of caspase in N-methyl-d-aspartate-Induced apoptosis in cerebrocortical neurons. J. Neurochem. 1998, 71, 946–959. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merino, J.J.; Garcimartín, A.; López-Oliva, M.E.; Benedí, J.; González, M.P. The Impact of CXCR4 Blockade on the Survival of Rat Brain Cortical Neurons. Int. J. Mol. Sci. 2016, 17, 2005. https://doi.org/10.3390/ijms17122005
Merino JJ, Garcimartín A, López-Oliva ME, Benedí J, González MP. The Impact of CXCR4 Blockade on the Survival of Rat Brain Cortical Neurons. International Journal of Molecular Sciences. 2016; 17(12):2005. https://doi.org/10.3390/ijms17122005
Chicago/Turabian StyleMerino, José Joaquín, Alba Garcimartín, María Elvira López-Oliva, Juana Benedí, and María Pilar González. 2016. "The Impact of CXCR4 Blockade on the Survival of Rat Brain Cortical Neurons" International Journal of Molecular Sciences 17, no. 12: 2005. https://doi.org/10.3390/ijms17122005
APA StyleMerino, J. J., Garcimartín, A., López-Oliva, M. E., Benedí, J., & González, M. P. (2016). The Impact of CXCR4 Blockade on the Survival of Rat Brain Cortical Neurons. International Journal of Molecular Sciences, 17(12), 2005. https://doi.org/10.3390/ijms17122005