
Effect of Lead (Pb) on Inflammatory Processes in the Brain

 , , and
, , and 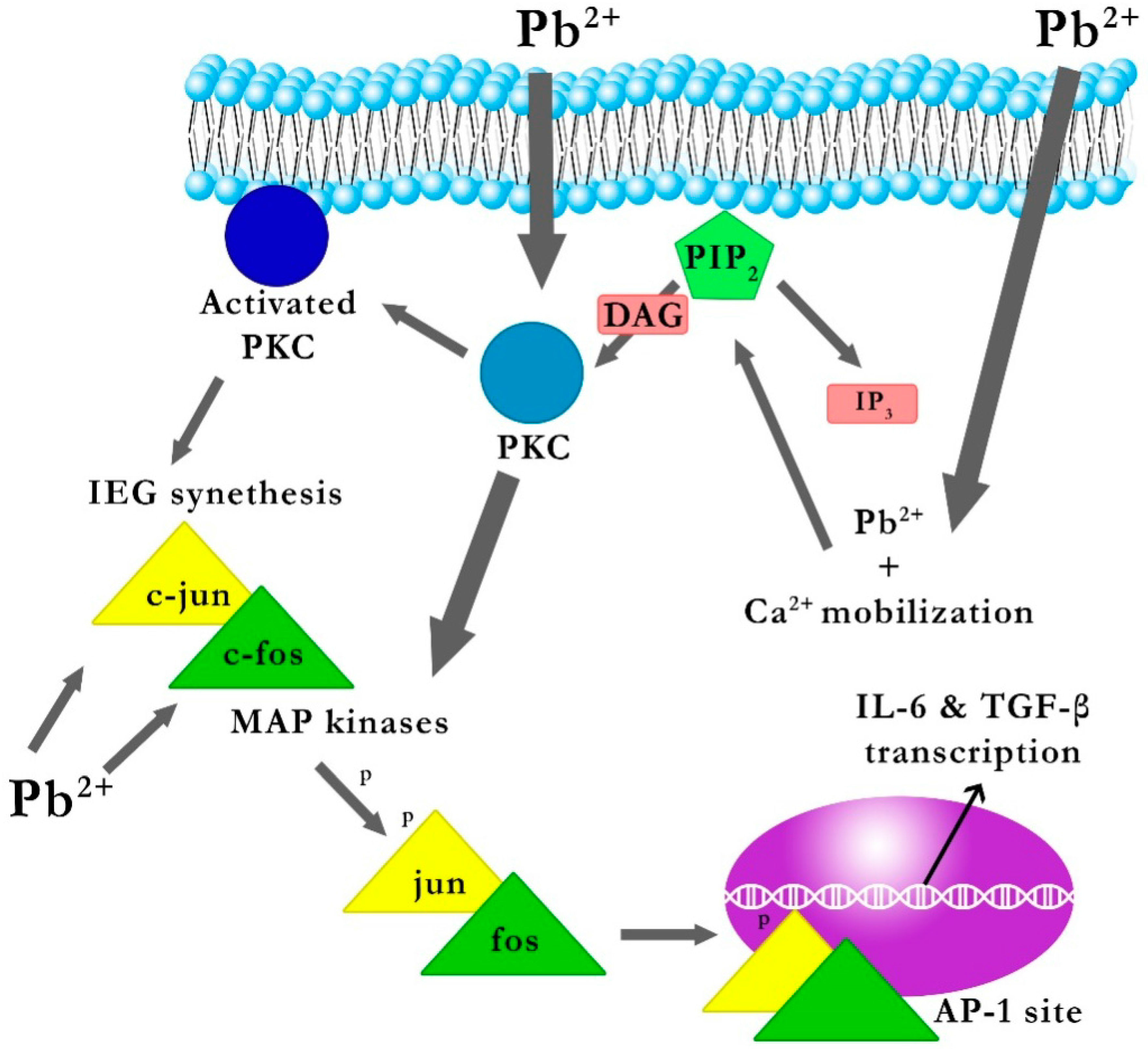{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effects of Lead on the Expression of Cytokines in Brain
2.1. Expression of Interleukin 6 and TGF-β1
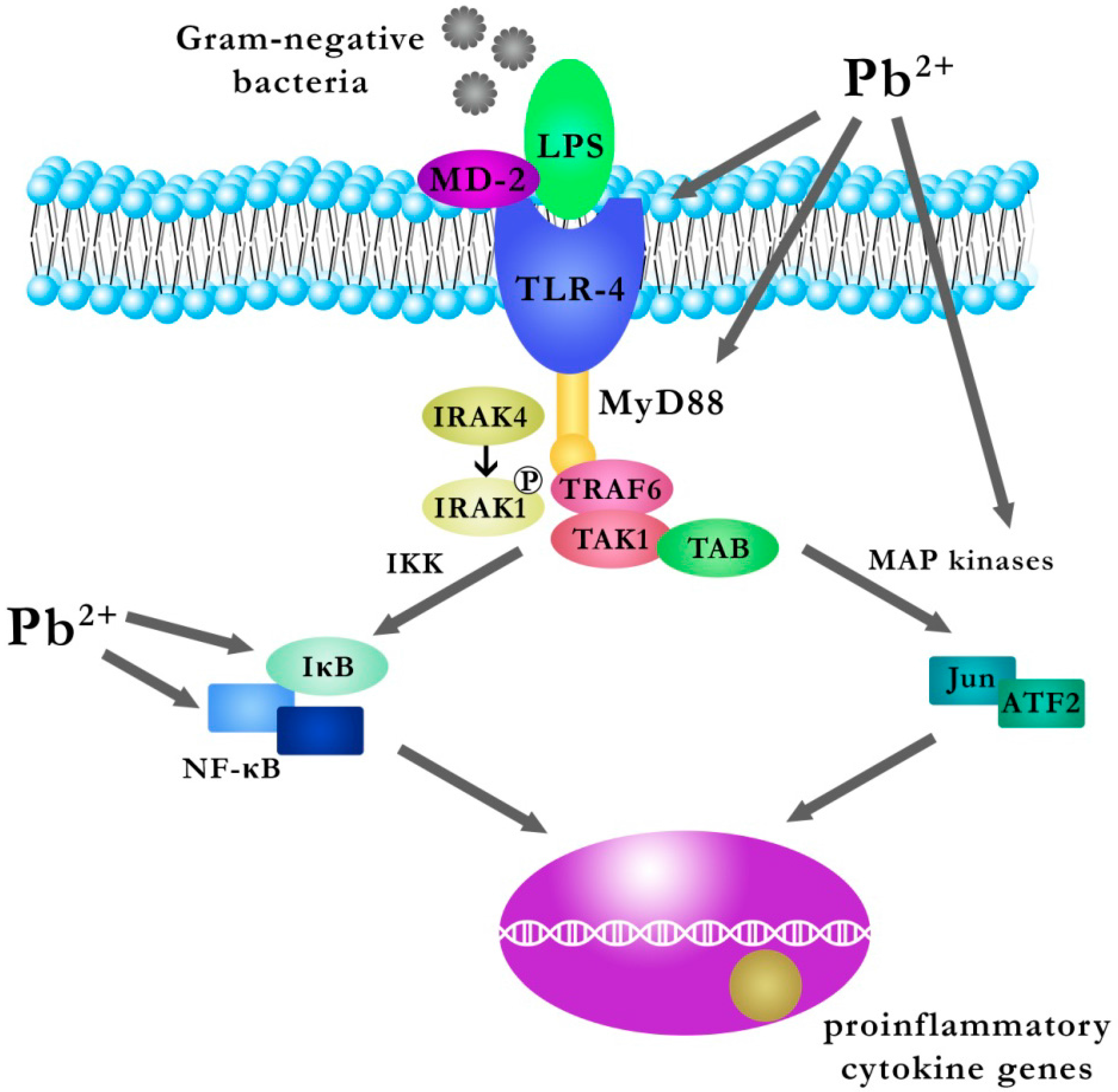
2.1.1. The Mechanism of the Effect of Pb on the Gene Expression of IL-6 and TGF-β1
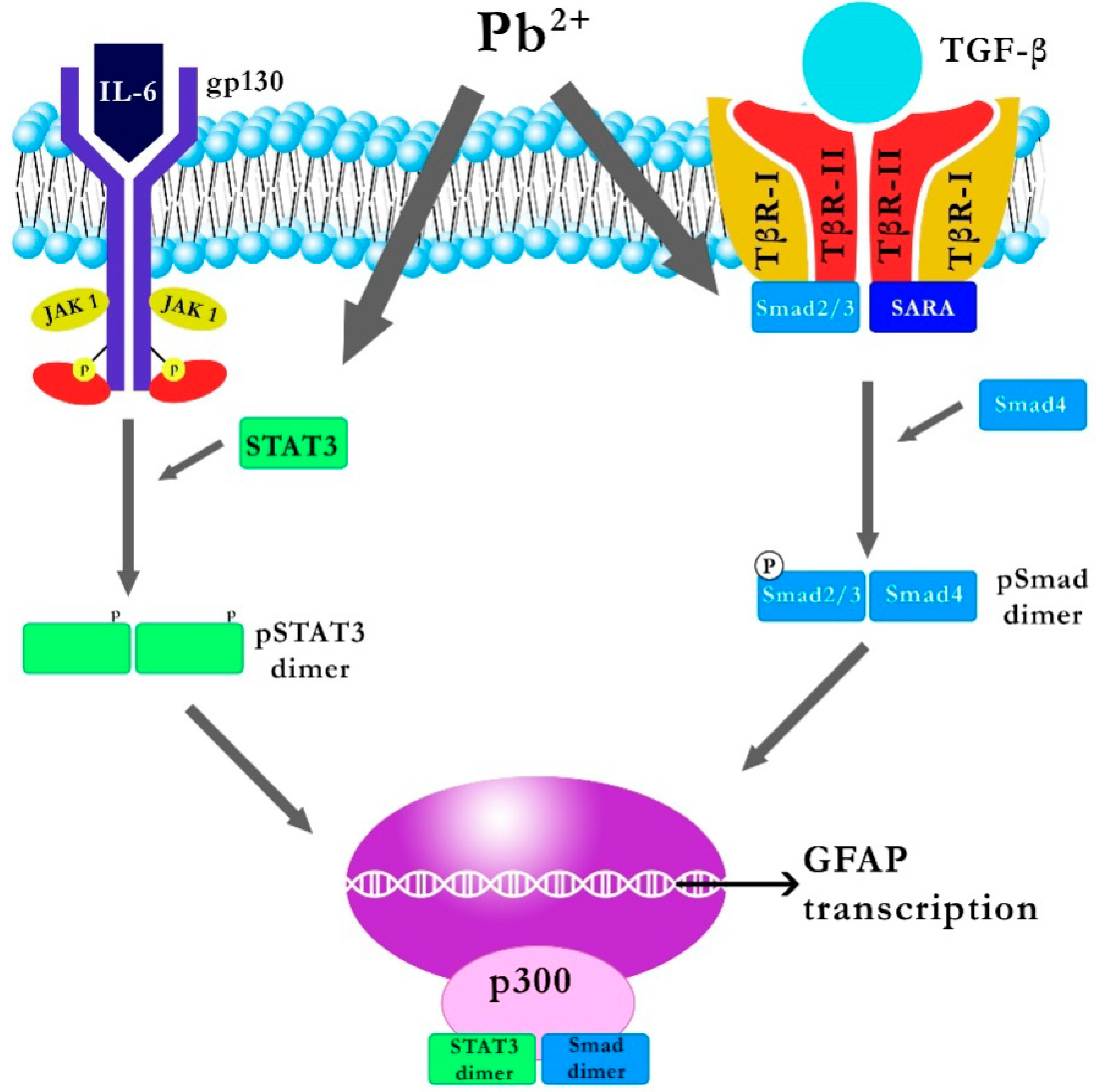
2.1.2. Effect of Pb on IL-6 and TGF-β1 Signal Transduction Pathways
2.2. Effect of Pb on Interleukin 16 Expression
2.3. Effect of Pb on Interleukin 18 Expression
2.4. Effect of Pb on Interleukin 10 Expression
3. Effects of Pb on Enzymes
3.1. Effects of Pb on the Gene Expression and Activity of Cyclooxygenase-2 (COX 2)
3.2. Effect of Pb on the Gene Expression and Activity of Caspase-1 and NOS 2
3.3. Effects of Pb on the Expression of Proteases
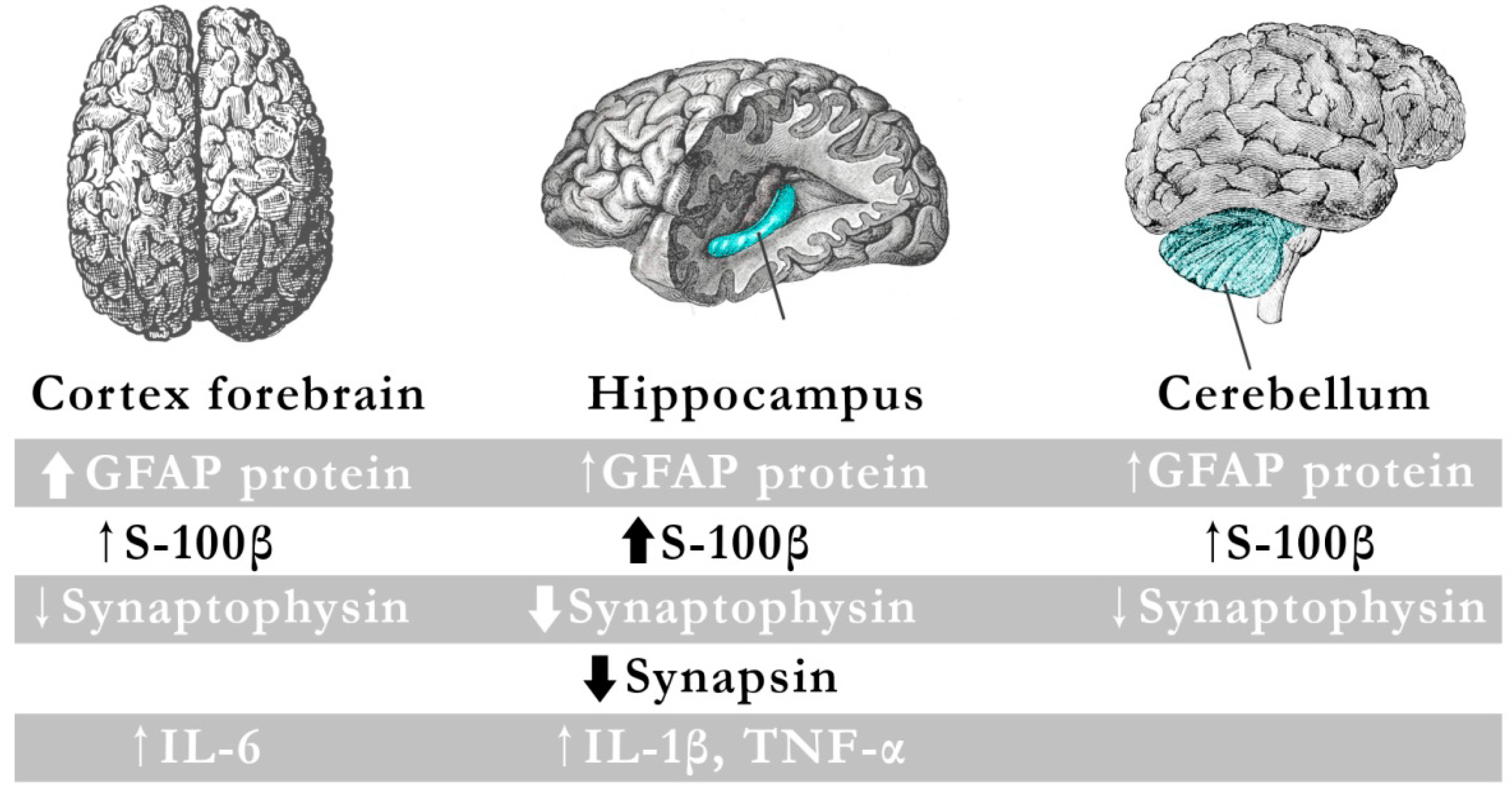
4. Effects of Pb on Microglia and Astroglia
5. Effects of Pb on the Expression of P2X4 and P2X7
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Steinman, L. Elaborate interactions between the immune and nervous systems. Nat. Immunol. 2004, 5, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Szelenyi, J.; Vizi, E.S. The catecholamines-cytokine balance: Interaction between the brain and the immune system. Ann. N. Y. Acad. Sci. 2007, 1113, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Pabello, N.; Bolivar, V.J.; Lawrence, D.A. Developmental lead effects on behavior and brain gene expression in male and female BALB/cAnNTac mice. Neurotoxicology 2012, 33, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Gonzales-Scarano, F.; Baltuch, G. Microglia as mediators of inflammatory and degenerative diseases. Annu. Rev. Neurosci. 1999, 22, 219–240. [Google Scholar] [CrossRef] [PubMed]
- Marx, F.; Blasko, I.; Grubeck-Loebenstein, B. Mechanisms of immune regulation in Alzheimer’s disease: A viewpoint. Arch. Immunol. Ther. Exp. 1999, 47, 205–209. [Google Scholar]
- McGeer, P.M.; Kawamata, T.; Walker, D.G.; Akijama, H.; Tooyama, I.; McGeer, E.G. Microglia in the degenerative neurological disease. Glia 1993, 71, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Mrak, R.E.; Griffin, W.S. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Bunn, T.L.; Parsons, P.J.; Kao, E.; Dietert, R.R. Exposure to lead during critical windows of embryonic development: Differential immunotoxic outcome based on stage of exposure and gender. Toxicol. Sci. 2001, 64, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Barton, H.J. Advantages of the use of deciduous teeth, hair, and blood analysis for lead and cadmium bio-monitoring in children. A study of 6-year-old children from Krakow (Poland). Biol. Trace Elem. Res. 2011, 143, 637–658. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Wright, J.M.; Rice, G.; Buckley, B.T.; Magsumbol, M.S.; Barr, D.B.; Williams, B.L. Metal exposures in an inner-city neonatal population. Environ. Int. 2010, 36, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Ning, Li.; Pingan, Z.; Mingwu, Q.; Jianfeng, S.; Haozhe, Li.; Wei, X. The effects of early life lead exposure on the expression of P2X7 receptor and synaptophysin in the hippocampus of mouse pups. J. Trace Elem. Med. Biol. 2015, 30, 124–128. [Google Scholar]
- Baranowska-Bosiacka, I.; Dąbrowska-Bouta, B.; Strużyńska, L. Regional changes in purines and selected purinergic receptors in immature rat brain exposed to lead. Toxicology 2011, 279, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.W. Brain capillaries: A target for inorganic lead poisoning. Neurotoxicology 1994, 5, 167–176. [Google Scholar]
- Goldstein, G.W. Developmental neurobiology of lead toxicity. In Human Lead Exposure, 1st ed.; Needleman, H.L., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 137–154. [Google Scholar]
- Kasten-Jolly, J.; Heo, Y.; Lawrence, D.A. Central Nervous System Cytokine Gene Expression: Modulation by Lead. J. Biochem. Mol. Toxicol. 2011, 25, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Canfield, R.L.; Gendle, M.H.; Cory-Slechta, D.A. Impaired neuropsychological functioning in lead-exposed children. Dev. Neuropsychol. 2004, 26, 513–540. [Google Scholar] [CrossRef] [PubMed]
- Winneke, G.; Krämer, U. Neurobehavioral aspects of lead neurotoxicity in children. Cent. Eur. J. Public Health. 1997, 5, 65–69. [Google Scholar] [PubMed]
- Lidsky, T.I.; Schneider, J.S. Lead neurotoxicity in children: Basic mechanisms and clinical correlates. Brain 2003, 126, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Strużynska, L.; Dabrowska-Bouta, B.; Koza, K.; Sulkowski, G. Inflammation-like glial response in lead-exposed immature rat brain. Toxicol. Sci. 2007, 95, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Boskabady, M.; Koohi, M.; Sadeghi-Hashjin, G.; Moin, M. The effect of lead exposure on selected blood inflammatory biomarkers in guinea pigs. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Sirivarasai, J.; Wananukul, W.; Kaojarem, S.; Chanprasertyothin, S.; Thongmung, N.; Ratanachaiwong, W.; Sura, T.; Sritara, P. Association between inflammatory marker, environmental lead exposure, and glutathione S-transferase gene. BioMed Res. Int. 2013, 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Oprica, M.; Eriksson, C.; Schulzberg, M. Inflammatory mechanisms associated with brain damage induced by kainic acid with special reference to the intereukin-1 system. J. Cell. Mol. Med. 2003, 7, 127–140. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.C.; Hu, S.; Peterson, P.K. Glia, cytokines, and neurotoxicity. Crit. Rev. Neurobiol. 1995, 9, 189–205. [Google Scholar] [PubMed]
- Liu, J.S.; John, G.R.; Sikora, A.; Lee, S.C.; Brosnan, C.F. Modulation of interleukin-1β and tumor necrosis factor α signaling by P2 purinergic receptors in human fetal astrocytes. J. Neurosci. 2000, 20, 5292–5299. [Google Scholar] [PubMed]
- Gruol, D.L.; Nelson, T.E. Physiological and pathological roles of interleukin-6 in the central nervous system. Mol. Neurobiol. 1997, 15, 307–339. [Google Scholar] [CrossRef] [PubMed]
- Van Wagoner, N.J.; Benveniste, E.N. Interleukin-6 expression and regulation in astrocytes. Neuroimmunology 1999, 100, 124–139. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Borrow, P.; Brooker, M.J.; Mucke, L. Astroglial overproduction of TGF-β1 enhances inflammatory central nervous system disease in transgenic mice. J. Neuroimmunol. 1997, 77, 45–50. [Google Scholar] [CrossRef]
- Bressler, J.; Kim, K.; Chakraborti, T.; Goldstein, G. Molecular mechanisms of lead neurotoxicity. Neurochem. Res. 1999, 24, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Long, G.J.; Rosen, J.F.; Schanne, F.A. Lead activation of protein kinase C from rat brain. J. Biol. Chem. 1994, 269, 834–837. [Google Scholar] [PubMed]
- Lu, H.; Guizzetti, M.; Costa, L. Inorganic lead activates the mitogen-activated protein kinase kinasemitogen-activated protein kinase-p90RSK signaling pathway in human astrocytoma cells via a protein kinase C-dependent mechanism. J. Pharmacol. Exp. Ther. 2002, 300, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ding, W.; Liu, X.; Mulder, K. c-Fos is required for TGFβ1 production and the associated paracrine migratory effects of human colon carcinoma cells. Mol. Carcinog. 2006, 45, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Persson, E.; Voznesensky, O.S.; Huang, Y.F.; Lerner, U.H. Increased expression of interleukin-6 by vasoactive intestinal peptide is associated with regulation of CREB, AP-1 and C/EBP, but not NF-κB, in mouse calvarial osteoblasts. Bone 2005, 37, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Brown, D.A.; Kitajima, I.; Xu, X.; Heidenreich, O.; Gryaznov, S.; Nerenberg, N. Binding and functional effects of transcriptional factor Sp1 on the murine interleukin-6 promotor. J. Biol. Chem. 1996, 271, 7330–7335. [Google Scholar] [PubMed]
- Kim, Y.; Ratziu, V.; Choi, S.G.; Lalazar, A.; Theiss, G.; Dang, Q.; Kim, S.J.; Friedman, S.L. Transcriptional activation of transforming growth factor β1 and its receptors by the Kruppel-like Zf9/core promoter-binding protein and Sp1. J. Biol. Chem. 1998, 273, 33750–33758. [Google Scholar] [CrossRef] [PubMed]
- Atkins, D.S.; Basha, M.R.; Zawia, N.H. Intracellular signaling pathways involved in mediating the effects of lead on the transcription factor Sp1. Int. J. Dev. Neurosci. 2003, 21, 235–244. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIFOSM-IL-6β receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Muller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signaling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Talar, B.; Czyż, M. TGF-β signaling pathways in cancers. Postepy Hig. Med. Dosw. 2013, 67, 1008–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGF-β signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Fukuda, S. Role of IL-6 in the neural stem cell differentiation. Clin. Rev. Allergy Immunol. 2005, 28, 249–256. [Google Scholar] [CrossRef]
- Kurschner, C.; Yuzaki, M. Neuronal interleukin-16 (NIL-16): A dual function PDZ domain protein. J. Neurosci. 1999, 19, 7770–7780. [Google Scholar] [PubMed]
- Wheeler, R.D.; Brough, D.; Le Feuvre, R.A.; Takeda, K.; Iwakura, Y.; Luheshi, G.N.; Rothwell, N.J. Interleukin-18 induces expression and release of cytokines from murine glial cells: Interactions with interleukin-1β. J. Neurochem. 2003, 85, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Conti, B.; Park, L.C.; Calingasan, N.Y.; Kim, Y.; Kim, H.; Bae, Y.; Gibson, G.E.; Joh, T.H. Cultures of astrocytes and microglia express interleukin 18. Brain Res. Mol. Brain Res. 1999, 67, 46–52. [Google Scholar] [CrossRef]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttila, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Curran, B.; O’Conner, J.J. The pro-inflammatory cytokine interleukin-18 impairs long-termpotentiation and NMDA receptor mediated transmission in the rat hippocampus in vitro. Neuroscience 2001, 108, 83–90. [Google Scholar] [CrossRef]
- Cordova, F.M.; Rodrigues, A.L.S.; Giacomelli, M.B.O.; Oliveira, C.S.; Posser, T.; Dunkley, P.R.; Leal, R.B. Lead stimulate ERK1/2 and p38MAPK phosphorylation in the hippocampus of immature rats. Brain Res. 2004, 998, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Bongiorno, P.B.; Rettori, V.; McCann, S.M.; Lici, J. Interleukin (IL) 1β, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in central nervous system and anterior pituitary during systemic inflammation with pathophysiological implications. Proc. Natl. Acad. Sci. USA 1997, 94, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Vane, J.R.; Bakhle, Y.S.; Botting, R.M. Cyclooxygenses 1 and 2. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 97–120. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Du, K.; Cai, Q.; Ma, L.; Jiao, Z.; Tan, J.; Xu, Z.; Li, J.; Luo, W.; Chen, J.; et al. Lead induces COX-2 expression in glial cells in a NFAT-dependent, AP-1/NFκB-independent manner. Toxicology 2014, 325, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, W.E.; Worley, P.F.; Pegg, J.; Bremer, M.; Isakson, P. COX-2, a synaptically induced enzyme, is expressed by excitatory neurons at postsynaptic sites in rat cerebral cortex. Proc. Natl. Acad. Sci. USA 1996, 3, 2317–2321. [Google Scholar] [CrossRef]
- Breder, C.; Smith, W.L.; Raz, A.; Masferrer, J.; Seibert, K.; Needleman, P.; Saper, C.B. Distribution and characterization of cyclooxygenase immunoreactivityin the ovine brain. J. Comp. Neurol. 1992, 322, 409–438. [Google Scholar] [CrossRef] [PubMed]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999, 830, 226–236. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 3, 901–910. [Google Scholar] [CrossRef]
- Minghetti, L. Role of COX-2 in inflammatory and degenerative brain diseases. Sub-Cell Biochem. 2007, 2, 127–141. [Google Scholar]
- Ding, J.; Li, J.; Xue, C.; Wu, K.; Ouyang, W.; Zhang, D.; Yan, Y.; Huang, C. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in beas-2B cells. J. Biol. Chem. 2006, 281, 24405–24413. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, J.; Wu, K.; Ouyang, W.; Ding, J.; Liu, Z.G.; Costa, M.; Huang, C. JNK1, but not JNK2, is required for COX-2 induction by nickelcompounds. Carcinogenesis 2006, 28, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.; Li, X.; Ding, J.; Luo, W.; Li, J.; Huang, C. A Cross-Talk Between NFAT and NF-κB Pathways is Crucial for Nickel- Induced COX-2 Expression in Beas-2B Cells. Curr. Cancer Drug Targets 2011, 11, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Zhang, D.; Ma, Q.; Li, J.; Huang, C. Cyclooxygenase-2 induction by arsenite through the IKKβ/NFκB pathway exerts an antiapoptotic effect in mouse epidermal Cl41 cells. Environ. Health Perspect. 2007, 115, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.; Ouyang, W.; Li, J.; Costa, M.; Huang, C. Cyclooxygenase-2 (COX-2) mediates arsenite inhibition of UVB-induced cellular apoptosis in mouse epidermal Cl41 cells. Curr. Cancer Drug Targets 2012, 12, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin 1 and interleukin 18 as mediators of inflammation and the aging process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [PubMed]
- Juttler, E.; Bonmann, E.; Spranger, M.; Kolb-Bachofen, V.; Suschek, C.V. A novel role of interleukin-1-converting enzyme in cytokine-mediated inducible nitric oxide synthase gene expression: Implications for neuroinflammatory diseases. Mol. Cell. Neurosci. 2007, 34, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, G.T.; Jadhav, A.L. Levels of protein kinase C and nitric oxide synthase activity in rats exposed to sub chronic low level lead. Mol. Cell. Biochem. 2001, 223, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Sifringer, M.; Stefovska, V.; Endesfelder, S.; Stahel, P.F.; Genz, K.; Dzietko, M.; Ikonomidou, C.; Felderhoff-Mueser, U. Activation of caspase-1 dependent interactions in developmental brain trauma. Neurobiol. Dis. 2007, 25, 614–622. [Google Scholar] [CrossRef] [PubMed]
- Bunnett, N.W. Protease-activated receptors: How proteases signal to cells to cause inflammation and pain. Semin. Thromb. Hemost. 2006, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Bosiacka, I.; Listos, J.; Gutowska, I.; Machoy-Mokrzyńska, A.; Kolasa-Wołosiuk, A.; Tarnowski, M.; Puchałowicz, K.; Prokopowicz, A.; Talarek, S.; Listos, P.; et al. Effects of perinatal exposure to lead (Pb) on purine receptor expression in the brain and gliosis in rats tolerant to morphine analgesia. Toxicology 2016, 339, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-T.; Chen, B.-Y.; Zhang, J.-Q.; Kuang, F.; Chen, L.-W. Lead exposure induced microgliosis and astrogliosis in hippocampus of young mice potentially by triggering TLR4-MyD88-NFκB signaling cascades. Toxicol. Lett. 2015, 239, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Rolls, A.; Shechter, R.; London, A.; Ziv, Y.; Ronen, A.; Levy, R.; Schwartz, M. Toll-like receptors modulate adult hippocampal neurogenesis. Nat. Cell Biol. 2007, 9, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Majewska, M.; Szczepanik, M. The role of toll-like receptors (TLR) in innate and adaptive immune responses and their function in immune response regulation. Postepy Hig. Med. Dosw. 2006, 60, 52–63. [Google Scholar]
- Akira, S.; Takeda, K.; Kaisho, T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Xu, D.; Brint, E.K.; O’Neill, L.A. Negative regulation of Toll-like receptor-mediated immune responses. Nat. Rev. Immunol. 2005, 5, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Hoshino, K. Myeloid differentiation factor 88-dependent and -independent pathways in toll-like receptor signaling. J. Infect. Dis. 2003, 187, S356–S363. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Yamamoto, M.; Takeda, K. Role of adapters in Toll-like receptor signaling. Biochem. Soc. Trans. 2003, 31, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Kicielińska, J.; Pajtasz-Piasecka, E. The role of IL-10 in the modulation of the immune response in normal conditions and the tumor environment. Postepy Hig. Med. Dosw. 2014, 68, 879–892. [Google Scholar] [CrossRef]
- Piotrowska, A.; Iżykowska, I.; Podhorska-Okołów, M.; Zabel, M.; Dzięgiel, P. The structure of NF-κB family proteins and their role in apoptosis. Postepy Hig. Med. Dosw. 2008, 62, 64–74. [Google Scholar]
- Sato, K. Effects of microglia on neurogenesis. Glia 2015, 63, 1394–1405. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, K.J.; Lammers, J.H.C.M.; Hoogendijk, E.M.G.; Kulig, B.M. Changes in regional brain GFAP levels and behavioral functioning following subchronic lead acetate exposure in adult rats. Neurotoxicology 1996, 17, 725–734. [Google Scholar] [PubMed]
- O’Callaghan, J.P. The use of glial fibrillary acidic protein in first-tier assessments of neurotoxicity. J. Am. Coll. Toxicol. 1999, 10, 719–726. [Google Scholar] [CrossRef]
- Norenberg, M.D. Reactive astrocytosis. In The Role of Glia in Neurotoxicity; Aschner, M., Kimelberg, H.K., Eds.; CRC Press: Boca Raton, FL, USA; New York, NY, USA, 1996; pp. 93–107. [Google Scholar]
- Norton, W.T.; Aquino, D.A.; Hozumi, I.; Chiu, F.C.; Brosnan, C.F.T.I. Quantitative aspects of reactive gliosis. A review. Neurochem. Res. 1992, 17, 877–885. [Google Scholar]
- Maglione, M.; Tress, O.; Haas, B.; Karram, K.; Trotter, J.; Willecke, K.; Kettenmann, H. Oligodendrocytes in mouse corpus callosum are coupled via gap junction channels formed by connexin47 and connexin32. Glia 2010, 58, 1104–1117. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. S100: A multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell Biol. 2001, 33, 637–668. [Google Scholar] [CrossRef]
- Koppal, T.; Lam, A.G.M.; Guo, L.; van Eldik, L.J. S100B proteins that lack one or both cysteine residues can induce inflammatory responses in astrocytes and microglia. Neurochem. Int. 2001, 39, 401–407. [Google Scholar] [CrossRef]
- Giulian, D.; Baker, T.J.; Shih, L.C.; Lachman, L.B. Interleukin 1 of the central nervous system is produced by ameboid microglia. J. Exp. Med. 1986, 164, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Herx, L.M.; Yong, V.W. Interleukine-1 β is required for the early evolution of reactive astrogliosis following CNS lesion. J. Neuropathol. Exp. Neurol. 2001, 60, 961–971. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Imaizumi, T.; Fujimoto, K.; Matsou, N.; Kimura, K.; Ciu, X.; Matsumiya, T.; Tanji, K.; Shibata, T.; Tamo, W.; et al. Synergistic stimulation, by tumor necrosis factor-α and interferon-γ, of fractalkine expression in human astrocytes. Neurosci. Lett. 2001, 303, 132–136. [Google Scholar] [CrossRef]
- Chapman, G.A.; Moores, K.; Harrison, D.; Campbell, C.A.; Stewart, B.; Strijbos, P.J. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 2000, 20, RC87. [Google Scholar] [PubMed]
- Mizuno, T.; Kawanokuchi, J.; Numata, K.; Suzumura, A. Production and neuroprotective functions of fractalkine in the central nervous system. Brain Res. 2003, 979, 65–70. [Google Scholar] [CrossRef]
- Zujovic, V.; Benavides, J.; Vige, X.; Carter, C.; Taupin, V. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. Glia 2000, 29, 305–315. [Google Scholar] [CrossRef]
- Zawia, N.H.; Harry, G.J. Developmental exposure to lead interferes with glial and neuronal differential gene expression in the rat cerebellum. Toxicol. Appl. Pharmacol. 1996, 138, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. P2X receptors in sensory neurones. Br. J. Anaesth. 2000, 84, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, S.O.; Brosnan, C.F.; Scemes, E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 2006, 26, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Swanson, R.A. P2X7 receptor mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [PubMed]
- Mingam, R.; de Smedt, V.; Amédée, T.; Bluthé, R.M.; Kelley, K.W.; Dantzer, R.; Layé, S. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1β in the murine brain. Brain Behav. Immun. 2008, 22, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Morelli, A.; Ferrari, D.; Bolognesi, G.; Rizzuto, R.; di Virgilio, F. Proapoptotic plasma membrane pore: P2X7 receptor. Drug Dev. Res. 2001, 52, 571–578. [Google Scholar] [CrossRef]
- Skaper, S.D.; Debetto, P.; Giusti, P. The P2X7 purinergic receptor: From physiology to neurological disorders. FASEB J. 2010, 24, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Sim, J.A.; Young, M.T.; Sung, H.Y.; Surprenant, A. Reanalysis of P2X7 receptor expression in rodent brain. J. Neurosci. 2004, 24, 6307–6314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Han, P.; Faltynek, C.R.; Jarvis, M.F.; Shieh, C.C. Functional expression of P2X7 receptors in nonneuronal cells of rat dorsal root ganglion. Brain Res. 2005, 1052, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Akter, S.; Akundi, R.S. The two-hit hypothesis for neuroinflammation: Role of exogenous ATP in modulating inflammation in the brain. Front. Cell. Neurosci. 2014, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Gandelman, M.; Peluffo, H.; Beckman, J.S.; Cassina, P.; Barbeito, L. Extracellular ATP and the P2X7 receptor in astrocyte-mediated motor neuron death: Implications for amyotrophic lateral sclerosis. J. Neuroinflamm. 2010, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Torre, I.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodríguez-Antigüedad, A.; Sánchez-Gómez, M.V.; et al. P2X(7) receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, M.; Díez-Zaera, M.; Sánchez-Nogueiro, J.; Gómez-Villafuertes, R.; Canals, J.M.; Alberch, J.; Miras-Portugal, M.T.; Lucas, J.J. Altered P2X7-receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist admin-istration. FASEB J. 2009, 23, 1893–1906. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.K.; McLarnon, J.G. Block of purinergic P2X(7) receptor is neuroprotective in an animal model of Alzheimer’s disease. Neuroreport 2008, 19, 1715–1719. [Google Scholar] [CrossRef] [PubMed]
- Skoczyńska, A.; Poręba, R.; Sieradz, A.; Andrzejak, R.; Sieradzka, U. The influence of lead and cadmium on immune system function. Med. Pr. 2002, 53, 259–264. [Google Scholar] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int. J. Mol. Sci. 2016, 17, 2140. https://doi.org/10.3390/ijms17122140
Chibowska K, Baranowska-Bosiacka I, Falkowska A, Gutowska I, Goschorska M, Chlubek D. Effect of Lead (Pb) on Inflammatory Processes in the Brain. International Journal of Molecular Sciences. 2016; 17(12):2140. https://doi.org/10.3390/ijms17122140
Chicago/Turabian StyleChibowska, Karina, Irena Baranowska-Bosiacka, Anna Falkowska, Izabela Gutowska, Marta Goschorska, and Dariusz Chlubek. 2016. "Effect of Lead (Pb) on Inflammatory Processes in the Brain" International Journal of Molecular Sciences 17, no. 12: 2140. https://doi.org/10.3390/ijms17122140
APA StyleChibowska, K., Baranowska-Bosiacka, I., Falkowska, A., Gutowska, I., Goschorska, M., & Chlubek, D. (2016). Effect of Lead (Pb) on Inflammatory Processes in the Brain. International Journal of Molecular Sciences, 17(12), 2140. https://doi.org/10.3390/ijms17122140