
Mesenchymal Stem Cells Ameliorated Glucolipotoxicity in HUVECs through TSG-6

Abstract
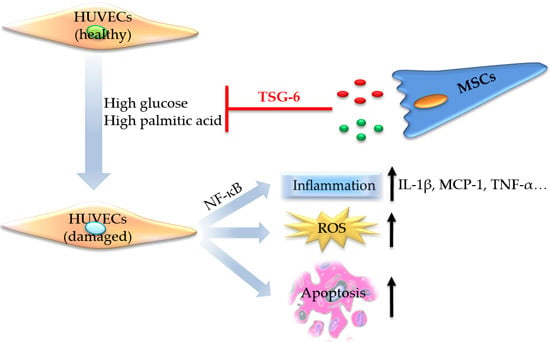
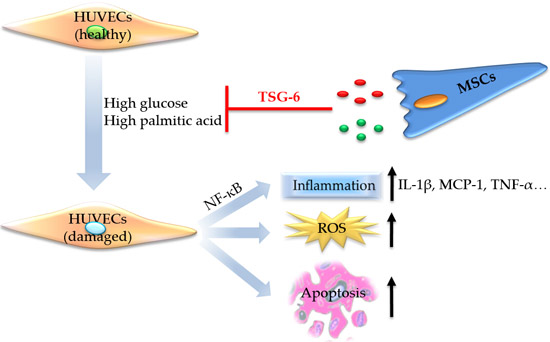
:
1. Introduction
2. Results
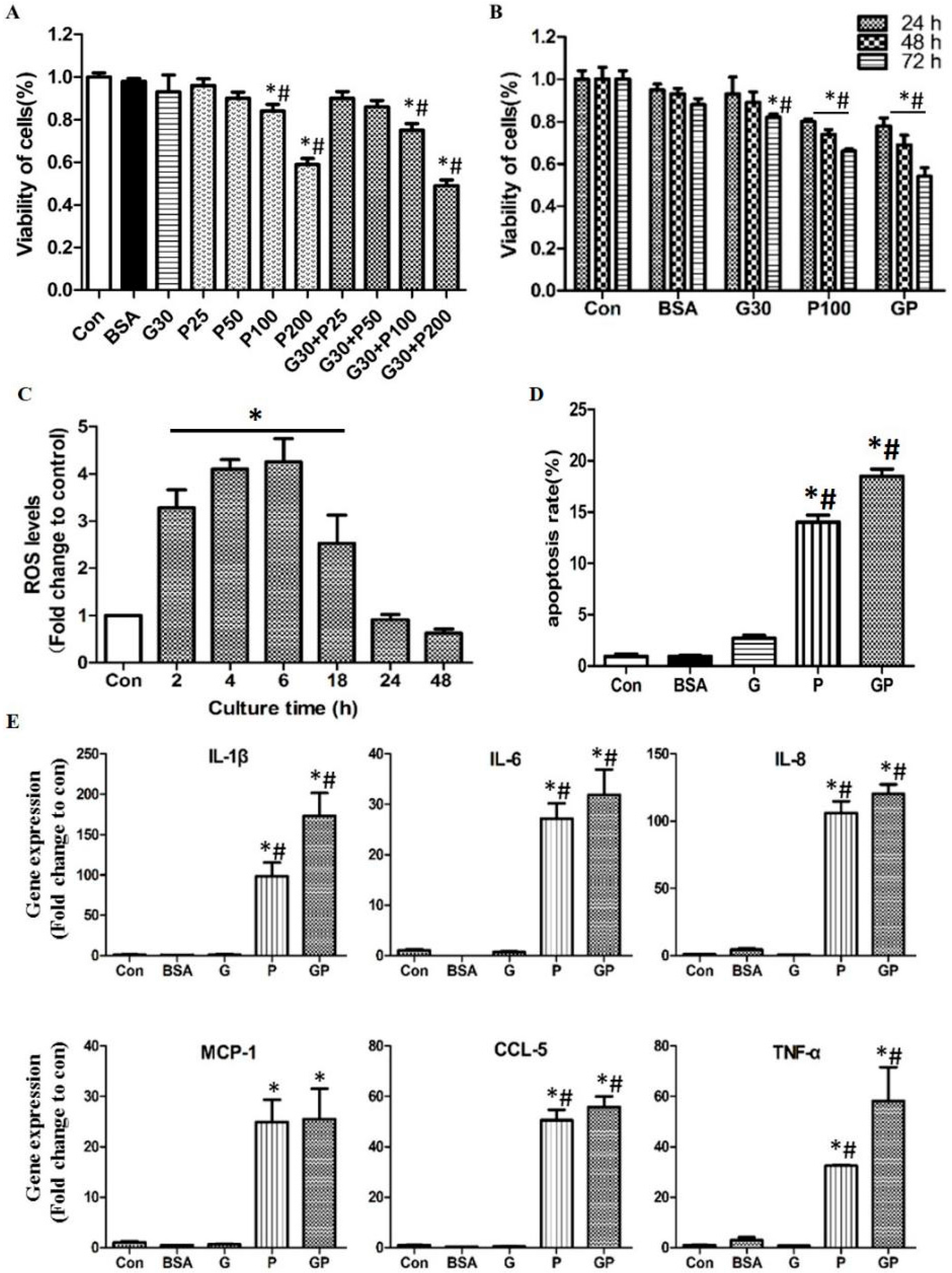
2.1. High Glucose and High Palmitic Acid Induced Inflammation and Cell Dysfunction in Human Umbilical Vein Endothelial Cells (HUVECs)
2.2. Protective Effects of Mesenchymal Stem Cells (MSCs) on HUVECs Dysfunction Driven by High Glucose and Palmitic Acid
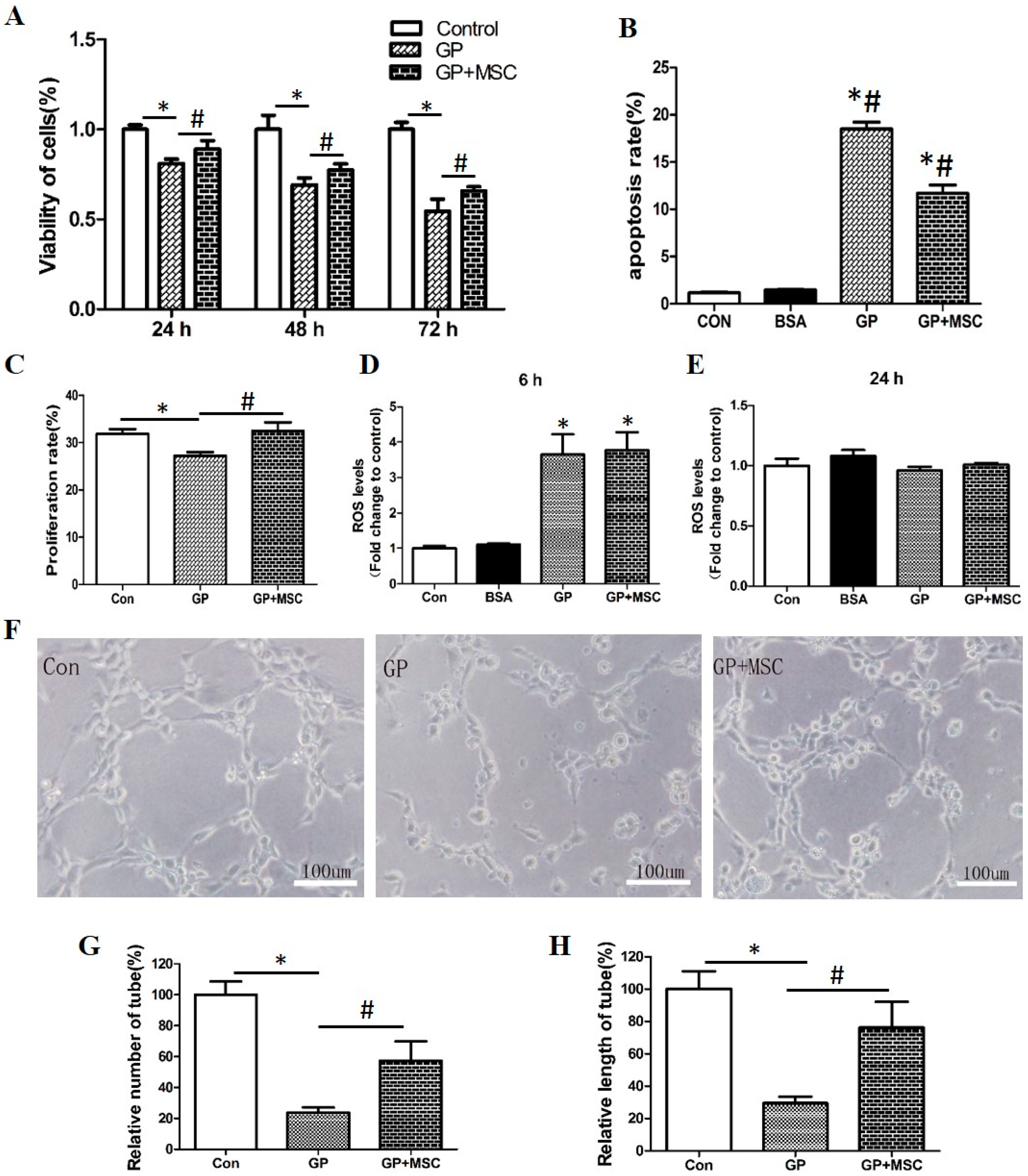
2.2.1. MSCs Alleviated Growth Arrest and Apoptosis but Not Reactive Oxygen Species (ROS) Production
2.2.2. MSCs Improved Capillary-Like Tube Formation Ability
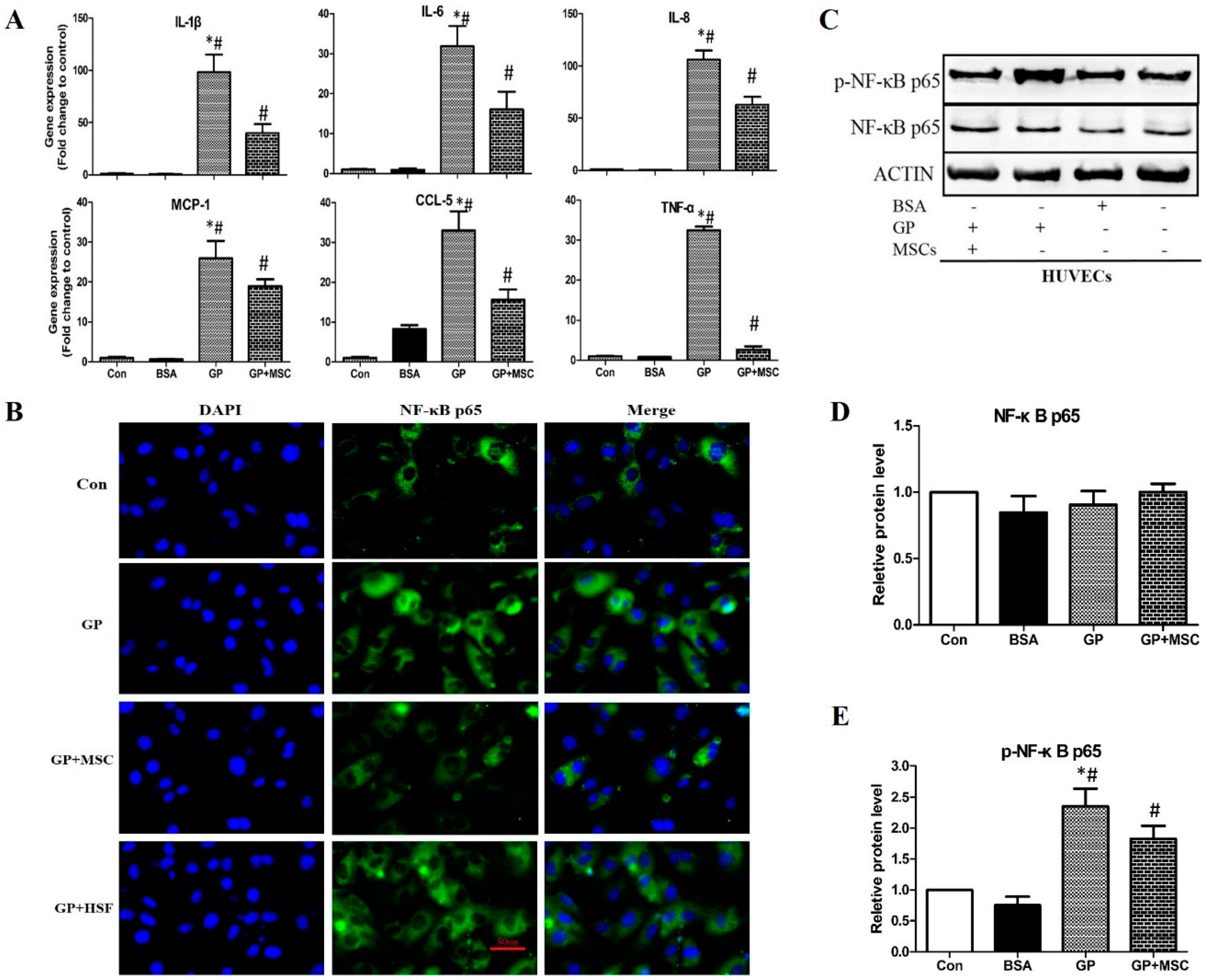
2.2.3. MSCs Reduced Gene Expression of Inflammatory Cytokines and Disrupted NF-κB Activation
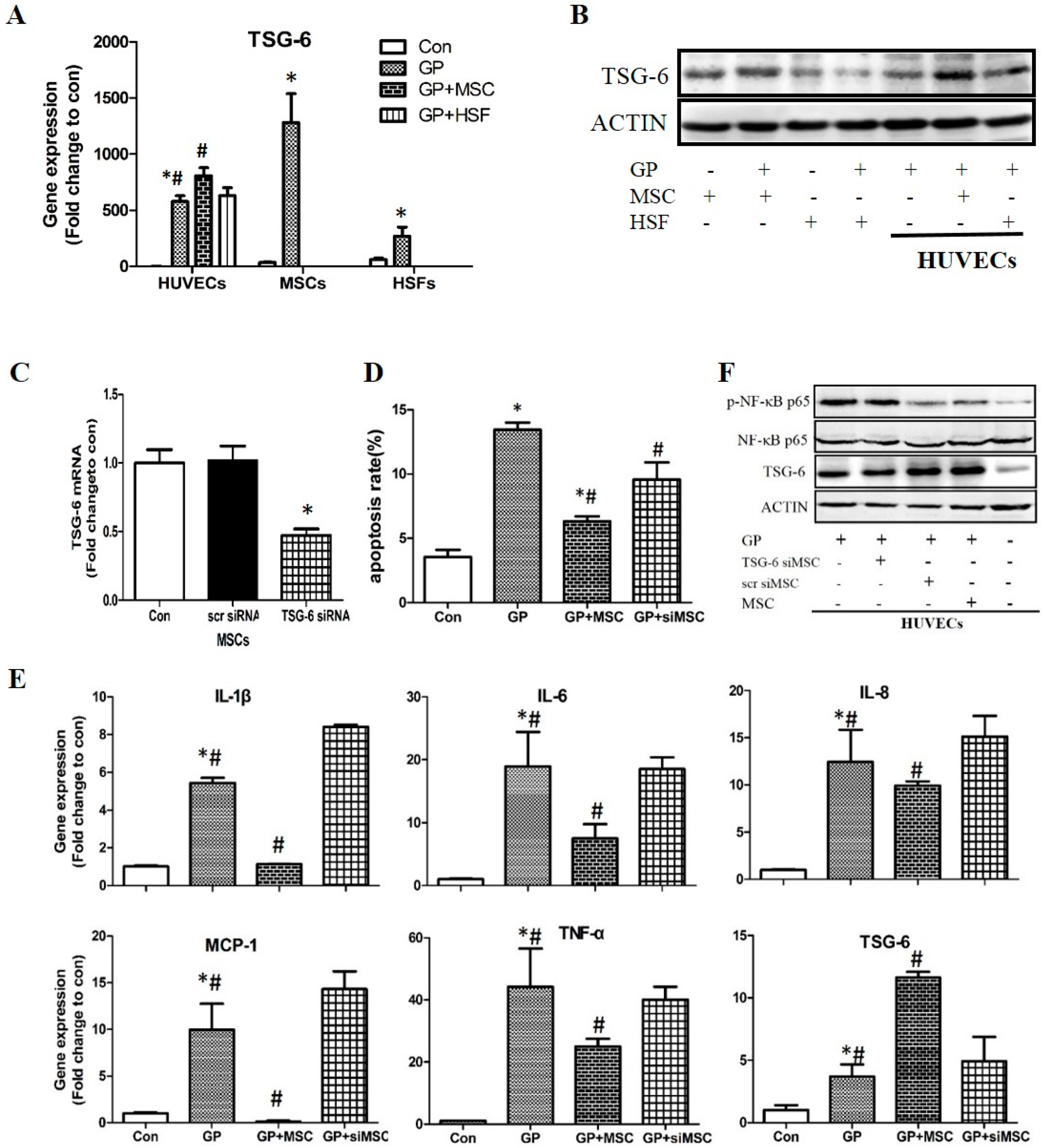
2.3. The Protective Role of MSCs Was Related to the Secretion of Tumor Necrosis Factor-α Stimulated Protein 6 (TSG-6)
2.3.1. Expression of TSG-6 in High Glucose and Palmitic Acid Stimulated Cells
2.3.2. The Effects of TSG-6 Knockdown on MSCs and MSCs Co-Cultured HUVECs
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Cultures
4.3. Assessment of Cell Viability
4.4. Flow Cytometry Analysis of ROS Production, Cell Cycle, and Apoptosis
4.5. Total RNA Extraction and Real-Time qPCR
4.6. Immunofluorescence Imaging
4.7. Endothelial Cell Capillary-Like Tube Formation Assay
4.8. Small Interfering RNAs (siRNAs) Transfection of MSCs
4.9. Western Blot Analysis
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mariana, M.; Barry, I.F.; John, S.P.; Peter, A.A.; Steven, C.E.; Ma, L.J. Lipotoxicity in diabetic nephropathy: The potential role of fatty acid oxidation. Clin. J. Am. Soc. Nephrol. 2010, 5, 2373–2379. [Google Scholar]
- Hideto, K.; Jongoh, K.; Lawrence, C. Emerging roles of hematopoietic cells in the pathobiology of diabetic complications. Trends Endocrinol. Metab. 2014, 25, 178–187. [Google Scholar]
- Dimitris, T.; Nikolaos, P.; Emmanuel, A.; Gerasimos, S.; George, L.; Konstantinos, T.; Christodoulos, S. Diabetes mellitus-associated vascular impairment—Novel circulating biomarkers and therapeutic approaches. J. Am. Coll. Cardiol. 2013, 62, 667–676. [Google Scholar]
- Sophia, Z.; Anushka, P.; John, C.; de Bastiaan, E.G.; Li, Q.; Laurent, B.; Mark, W.; Toshiharu, N.; Bruce, N.; Stephen, M.; et al. Severe hypoglycemia and risks of vascular events and death. N. Engl. J. Med. 2010, 363, 1410–1418. [Google Scholar]
- Francis, K.; Kelly, A.T.; Julie, R.; Matilda, P.; Lutfiyah, H.; Byron, M.G.; Arnold, S.B.; Pathmaja, P.; Cecilia, M.G.; Marshall, A.C.; et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKβ. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 989–994. [Google Scholar]
- Mittendorfer, B.; Liem, O.; Patterson, B.W.; Miles, J.M.; Klein, S. What does the measurement of whole-body fatty acid rate of appearance in plasma by using a fatty acid tracer really mean? Diabetes 2003, 52, 1641–1648. [Google Scholar] [CrossRef] [PubMed]
- Wende, A.R.; Symons, J.D.; Abel, E.D. Mechanisms of lipotoxicity in the cardiovascular system. Curr. Hypertens. Rep. 2012, 14, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Poitout, V. β-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 2004, 53, S119–S124. [Google Scholar] [CrossRef] [PubMed]
- Im, G.I.; Shin, Y.W.; Lee, K.B. Do adipose tissue-derived mesenchymal stem cells have the same osteogenic and chondrogenic potential as bone marrow-derived cells? Osteoarthr. Cartil. 2005, 13, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Campagnoli, C.; Roberts, I.A.; Kumar, S.; Bennett, P.R.; Bellantuono, I.; Fisk, N.M. Identification of mesenchymal stem/progenitor cells in human first-trimester fetal blood, liver, and bone marrow. Blood 2001, 98, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Keating, A. Mesenchymal stromal cells: New directions. Cell Stem Cell 2012, 10, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Nora, G.S.; Arnold, I.C. Mesenchymal stem cells: Mechanisms of inflammation. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 457–478. [Google Scholar]
- Davey, G.C.; Patil, S.B.; O’Loughlin, A.; O’Brien, T. Mesenchymal stem cell-based treatment for microvascular and secondary complications of diabetes mellitus. Front. Endocrinol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mazzucato, C.A.; Weir, S.B. Stem cell therapy for type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2010, 6, 139–148. [Google Scholar] [CrossRef] [PubMed]
- An, X.X.; Shi, M.M.; Yuan, Y.J.; Liu, J.P.; Chen, Y.N.; Chen, B.; Li, L.; Liao, G.N.; Cheng, J.Q.; Lu, Y.R.; et al. Intravenous injection of human mesenchymal stem cells ameliorated early renal damage in diabetic rhesus monkey model. In Proceedings of the IPITA-IXA-CTS Joint Congress, Melbourne, Australia, 15–19 November 2015.
- Ezekiel, M.; Ian, R.S.; David, M.H.; Matilda, P.; Norma, O.R.; Sanshiro, T.; Priya, H.; Michael, W.S.; Francis, K. Activation of NF-κB by palmitate in endothelial cells a key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1370–1375. [Google Scholar]
- Lee, R.H.; Pulin, A.A.; Seo, M.J.; Kota, D.J.; Ylostalo, J.; Larson, B.L.; Prieto, L.S.; Delafontaine, P.; Prockop, D.J. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009, 5, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Keane, K.; Newsholme, P. Metabolic regulation of insulin secretion. Vitam. Horm. 2014, 95, 1–33. [Google Scholar] [PubMed]
- Newsholme, P.; Krause, M. Nutritional regulation of insulin secretion: Implications for diabetes. Clin. Biochem. Rev. 2012, 33, 35–47. [Google Scholar] [PubMed]
- Kusminski, C.M.; Shetty, S.; Orci, L.; Unger, R.H.; Scherer, P.E. Diabetes and apoptosis: Lipotoxicity. Apoptosis 2009, 14, 1484–1495. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.J.; Shi, M.M.; Li, L.; Liu, J.P.; Chen, B.; Chen, Y.M.; An, X.X.; Liu, S.Y.; Luo, R.X.; Cheng, J.Q.; et al. Mesenchymal stem cell-conditioned media ameliorate high glucose-induced endothelial cell injury by improving mitochondrial bioenergetics via Sirt1/AMPK/PGC-1α Pathway. Clin. Sci. 2016, in press. [Google Scholar]
- Charlotte, J.G.; Katherine, M.; Sarah, F.; Hardie, D.G.; Kei, S.; Harinder, S.H. Counter-modulation of fatty acid-induced pro-inflammatory nuclear factor κB signaling in rat skeletal muscle cells by AMP-activated protein kinase. Biochem. J. 2011, 435, 463–474. [Google Scholar]
- Listenberger, L.L.; Ory, D.S.; Schaffer, J.E. Palmitate-induced apoptosis can occur through a ceramide-Independent Pathway. J. Biol. Chem. 2001, 276, 14890–14895. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Zhang, H.; Liu, J.; Chi, W.L.; Liu, P. Cyclooxygenase-2-dependent oxidative stress mediates palmitate-induced impairment of endothelium-dependent relaxations in mouse arteries. Biochem. Pharmacol. 2014, 91, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Piro, S.; Anello, M.; Di, P.C.; Lizzio, M.N.; Patane, G.; Rabuazzo, A.M.; Vigneri, R.; Purrello, M.; Purrello, F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: Possible role of oxidative stress. Metabolism 2002, 51, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef] [PubMed]
- Kharroubi, I.; Ladriere, L.; Cardozo, A.K.; Dogusan, Z.; Cnop, M.; Eizirik, D.L. Free fatty acids and cytokines induce pancreatic β-cell apoptosis by different mechanisms: Role of nuclear factor-κB and endoplasmic reticulum stress. Endocrinology 2004, 145, 5087–5096. [Google Scholar] [CrossRef] [PubMed]
- Laybutt, D.R.; Preston, A.M.; Akerfeldt, M.C.; Kench, J.G.; Busch, A.K.; Biankin, A.V.; Biden, T.J. Endoplasmic reticulum stress contributes to β cell apoptosis in type 2 diabetes. Diabetologia 2007, 50, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Lupi, R.; Dotta, F.; Marselli, L.; Masini, M.; Silvia, D.G.; Santangelo, C.; Patane, G.; Boggi, U.; Piro, S.; Anello, M.; et al. Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: Evidence that β-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 2002, 51, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Krishanthi, G.; Christopher, V.; Ross, B.; Chris, T.; Gustavo, D. Mechanisms of palmitate-induced cell death in human Osteoblasts. Biol. Open 2013, 2, 1382–1389. [Google Scholar]
- Swati, J.B.; Shirish, S.B.; Kiranmayi, A.; Leila, G.; Daniell, H.; Matthew, C.; Prachi, H.; Craig, J.M. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar]
- Harald, S.; Katrin, S.; Norbert, S.; Hans, G.W.; Fausto, M.; Monika, K.; Hans-Ulrich, H. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes 2004, 53, 3209–3216. [Google Scholar]
- Hajra, L.; Evans, A.I.; Chen, M.; Hyduk, S.J.; Collins, T.; Cybulsky, M.I. The NF-κB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc. Natl. Acad. Sci. USA 2000, 97, 9052–9057. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Yu, J.M.; Foskett, A.M.; Peltier, G.; Reneau, J.C.; Bazhanov, N.; Oh, J.Y.; Prockop, D.J. TSG-6 as a biomarker to predict efficacy of human mesenchymal stem/progenitor cells (hMSCs) in modulating sterile inflammation in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 16766–16771. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Yiu, W.H.; Li, R.X.; Wong, D.W.; Leung, J.C.; Chan, L.Y.; Zhang, Y.; Lian, Q.; Lin, M.; Tse, H.F.; et al. Mesenchymal stem cells modulatealbumin-induced renal tubular inflammation and fibrosis. PLoS ONE 2014, 9, e90883. [Google Scholar]
- Mindrescu, C.; Dias, A.A.; Olszewski, R.J.; Klein, M.J.; Reis, L.F.L.; Wisniewski, H.G. Reduced susceptibility to collagen-induced arthritis in DBA/1J mice expressing the TSG-6 transgene. Arthritis Rheumatol. 2002, 46, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- Sandor, S.; Tamas, B.; Istvan, G.; Tibor, T.G.; Katalin, M. Enhanced neutrophil extravasation and rapid progression of proteoglycan-induced arthritis in TSG-6-knockout mice. Arthritis Rheumatol. 2004, 50, 3012–3022. [Google Scholar]
- Choi, H.; Lee, R.H.; Bazhanov, N.; Oh, J.Y.; Prockop, D.J. Anti-inflammatory protein TSG-6 secreted by activated MSCs attenuates zymosan-induced mouse peritonitis by decreasing TLR2/NF-κB signaling in resident macrophages. Blood 2011, 118, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Hoak, J.C. An improved method for the addition of long-chain free fatty acid to protein solutions. Anal. Biochem. 1969, 32, 297–302. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| IL-1β | GCCGTGTCAGTTGTTGTAGC | TGAAGGGAATCAAGGTGCTC |
| IL-6 | TACATCCTCGACGGCATCTC | GCCATCTTTGGAAGGTTCAG |
| IL-8 | ACTCCAAACCTTTCCACCC | AACTTCTCCACAACCCTCTGC |
| MCP-1 | AGCCAGATGCAATCAATGCC | GGGTCAGCACAGATCTCCTT |
| CCL-5 | CCTGCTGCTTTGCCTACATT | GCACACACTTGGCGATTCT |
| TNF-α | CTGCCTGCTGCACTTTGGA | TTGAAGAGGACCTGGGAGTAGAT |
| TSG-6 | TGGCTTTGTGGGAAGATACTGT | TGGAAACCTCCAGCTGTCAC |
| GAPDH | GAACGGGAAGCTCACTGG | GCCTGCTTCACCACCTTCT |
| β-Actin | CCACGAAACTACCTTCAACTCC | GTGATCTCCTTCTGCATCCTGT |
| Gene | Target | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|---|
| si-h-TNFAIP6001 | GCAAATACAAGCTCACCTA | GCAAAUACAAGCUCACCUA | dTdT |
| dTdT | CGUUUAUGUUCGAGUGGAU | ||
| si-h-TNFAIP6002 | GCTACAACCCACACGCAAA | GCUACAACCCACACGCAAA | dTdT |
| dTdT | CGAUGUUGGGUGUGCGUUU | ||
| si-h-TNFAIP6003 | CCAGGTTGCTTGGCTGATT | CCAGGUUGCUUGGCUGAUU | dTdT |
| dTdT | GGUCCAACGAACCGACUAA |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, X.; Li, L.; Chen, Y.; Luo, A.; Ni, Z.; Liu, J.; Yuan, Y.; Shi, M.; Chen, B.; Long, D.; et al. Mesenchymal Stem Cells Ameliorated Glucolipotoxicity in HUVECs through TSG-6. Int. J. Mol. Sci. 2016, 17, 483. https://doi.org/10.3390/ijms17040483
An X, Li L, Chen Y, Luo A, Ni Z, Liu J, Yuan Y, Shi M, Chen B, Long D, et al. Mesenchymal Stem Cells Ameliorated Glucolipotoxicity in HUVECs through TSG-6. International Journal of Molecular Sciences. 2016; 17(4):483. https://doi.org/10.3390/ijms17040483
Chicago/Turabian StyleAn, Xingxing, Lan Li, Younan Chen, Ai Luo, Zuyao Ni, Jingping Liu, Yujia Yuan, Meimei Shi, Bo Chen, Dan Long, and et al. 2016. "Mesenchymal Stem Cells Ameliorated Glucolipotoxicity in HUVECs through TSG-6" International Journal of Molecular Sciences 17, no. 4: 483. https://doi.org/10.3390/ijms17040483
APA StyleAn, X., Li, L., Chen, Y., Luo, A., Ni, Z., Liu, J., Yuan, Y., Shi, M., Chen, B., Long, D., Cheng, J., & Lu, Y. (2016). Mesenchymal Stem Cells Ameliorated Glucolipotoxicity in HUVECs through TSG-6. International Journal of Molecular Sciences, 17(4), 483. https://doi.org/10.3390/ijms17040483