
The Molecular Epidemiology and Evolutionary Dynamics of Influenza B Virus in Two Italian Regions during 2010–2015: The Experience of Sicily and Liguria

 , ,
, ,  ,
,
Abstract
:1. Introduction
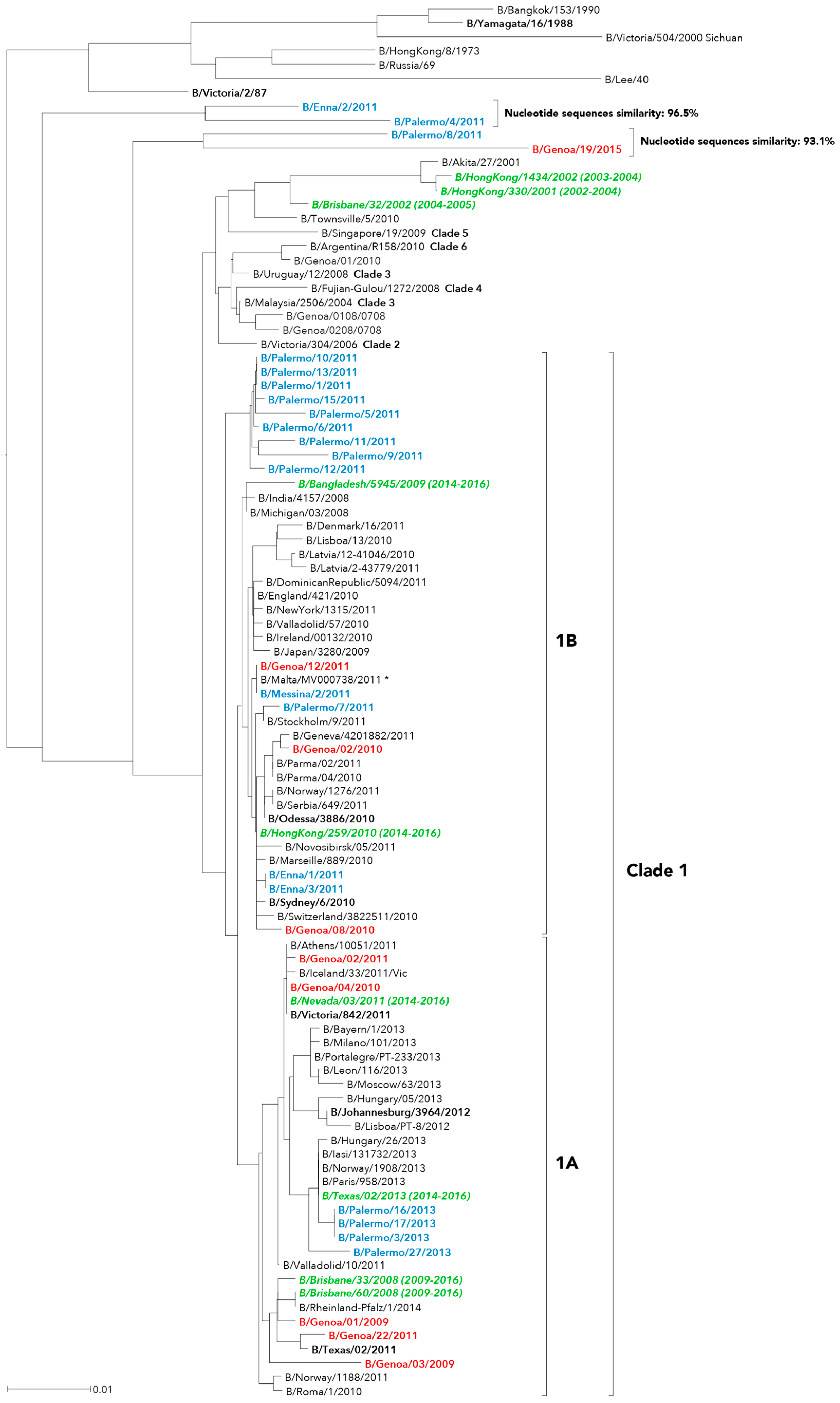
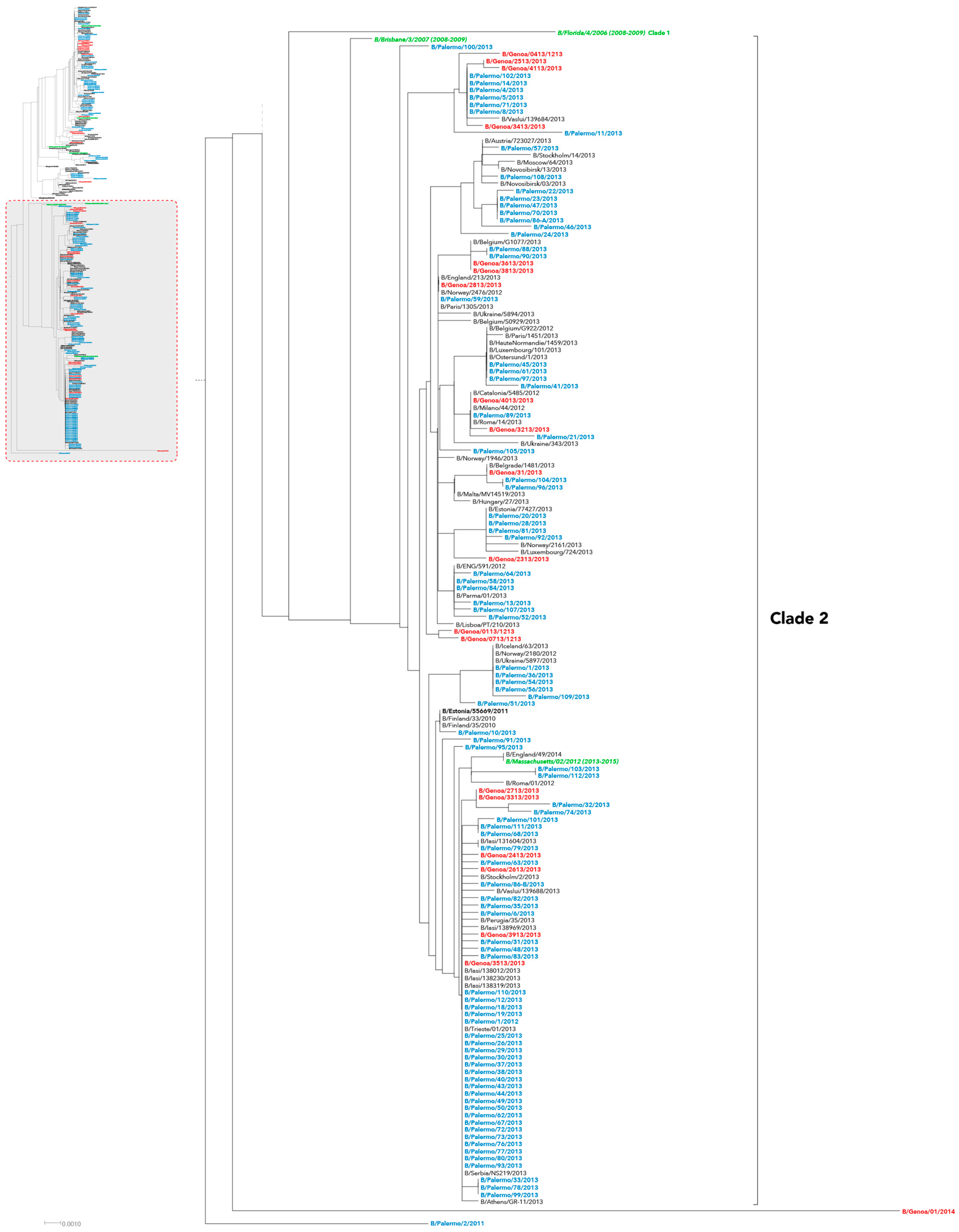
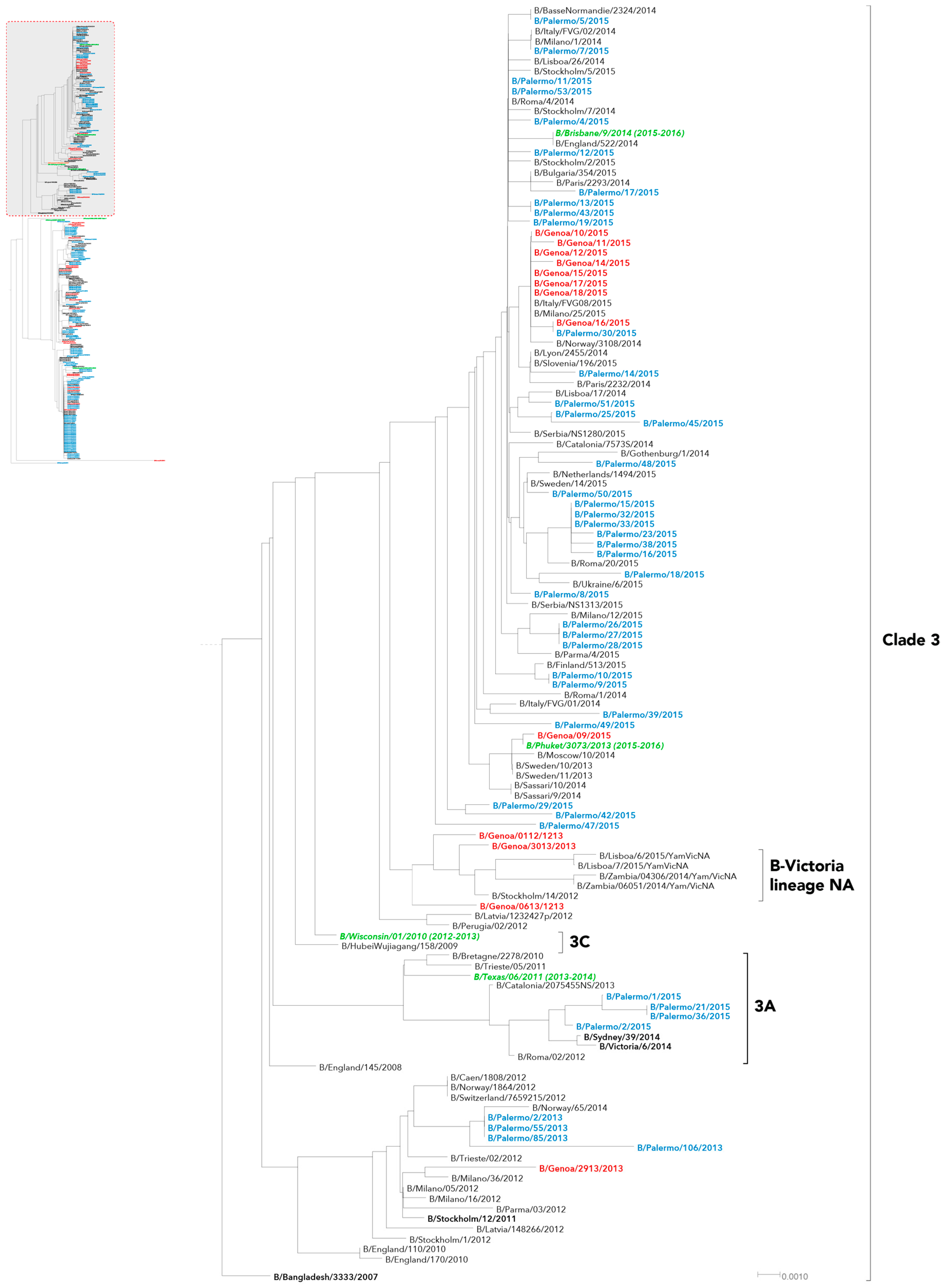
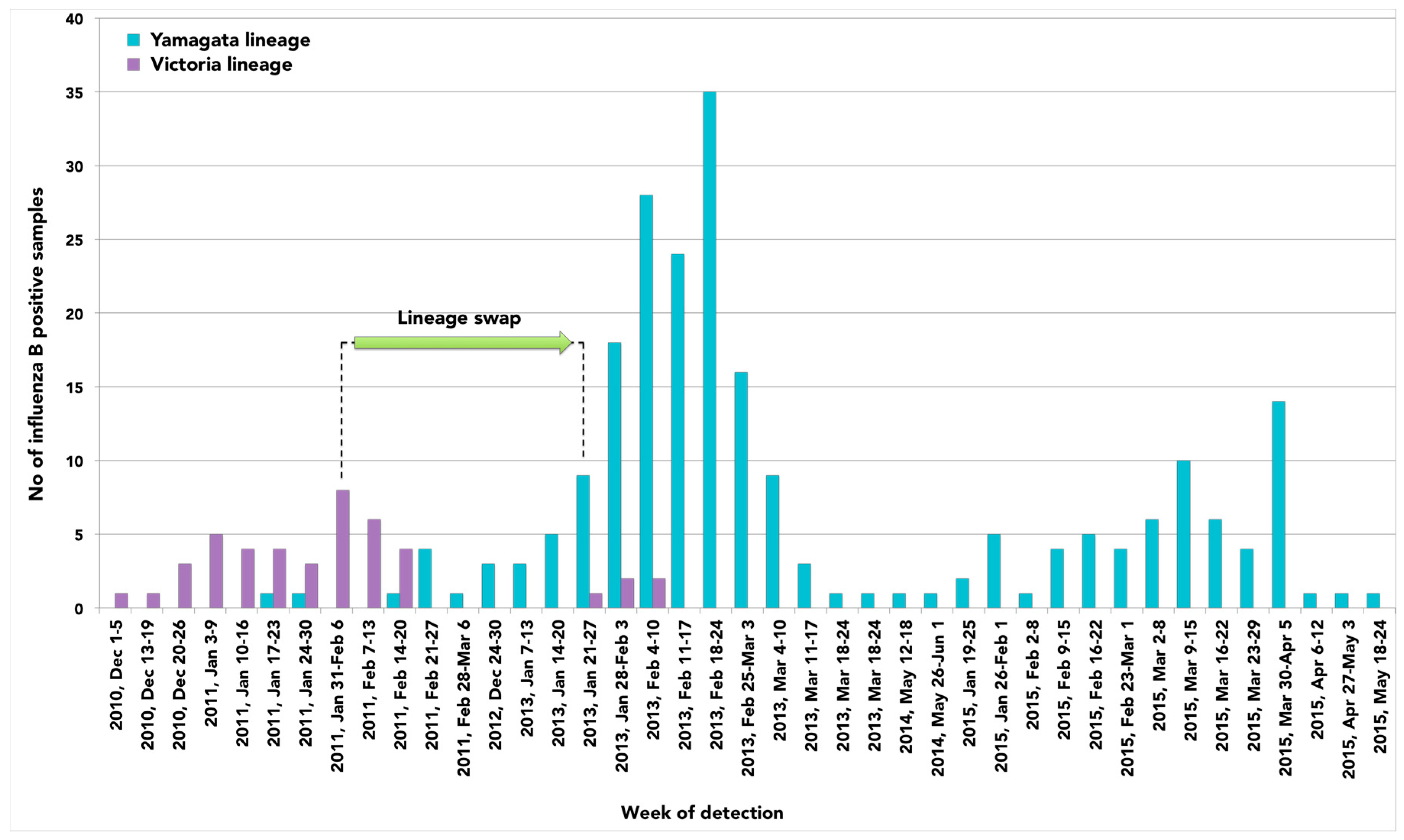
2. Results
3. Discussion
4. Materials and Methods
4.1. Study Population and Clinical Specimens Collection
4.2. Phylogenetic Analysis and Antigenic Characterization
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| HA | Hemagglutinin |
| ILI | Influenza-like illness |
References
- Fraaij, P.L.; Heikkinen, T. Seasonal influenza: The burden of disease in children. Vaccine 2001, 29, 7524–7528. [Google Scholar] [CrossRef] [PubMed]
- Paul Glezen, W.; Schmier, J.K.; Kuehn, C.M.; Ryan, K.J.; Oxford, J. The burden of influenza B: A structured literature review. Am. J. Public Health 2013, 103, e43–e51. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.; Chaves, S.S.; Daily Kirley, P.; Emerson, R.; Aragon, D.; Hancock, E.B.; Butler, L.; Baumbach, J.; Hollick, G.; Bennett, N.M.; et al. Estimating influenza disease burden from population-based surveillance data in the United States. PLoS ONE 2015, 10, e0118369. [Google Scholar]
- Osterhaus, A.D.; Rimmelzwaan, G.F.; Martina, B.E.; Bestebroer, T.M.; Fouchier, R.A. Influenza B virus in seals. Science 2000, 288, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Bodewes, R.; Morick, D.; de Mutsert, G.; Osinga, N.; Bestebroer, T.; van der Vliet, S.; Smits, S.L.; Kuiken, T.; Rimmelzwaan, G.F.; Fouchier, R.A.; et al. Recurring influenza B virus infections in seals. Emerg. Infect. Dis. 2013, 19, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Ran, Z.; Shen, H.; Lang, Y.; Kolb, E.A.; Turan, N.; Zhu, L.; Ma, J.; Bawa, B.; Liu, Q.; Liu, H.; et al. Domestic pigs are susceptible to infection with influenza B viruses. J. Virol. 2015, 89, 4818–4826. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, Y.; Sugawara, K.; Takashita, E.; Muraki, Y.; Hongo, S.; Katsushima, N.; Mizuta, K.; Nishimura, H. Genetic diversity of influenza B virus: The frequent reassortment and cocirculation of the genetically distinct reassortant viruses in a community. J. Méd. Virol. 2004, 74, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Nerome, R.; Hiromoto, Y.; Sugita, S.; Tanabe, N.; Ishida, M.; Matsumoto, M.; Lindstrom, S.E.; Takahashi, T.; Nerome, K. Evolutionary characteristics of influenza B virus since its first isolation in 1940: Dynamic circulation of deletion and insertion mechanism. Arch. Virol. 1998, 143, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Kanegae, Y.; Sugita, S.; Endo, A.; Ishida, M.; Senya, S.; Osako, K.; Nerome, K.; Oya, A. Evolutionary pattern of the hemagglutinin gene of influenza B viruses isolated in Japan: Cocirculating lineages in the same epidemic season. J. Virol. 1990, 64, 2860–2865. [Google Scholar] [PubMed]
- Lin, Y.P.; Gregory, V.; Bennett, M.; Hay, A. Recent changes among human influenza viruses. Virus Res. 2004, 103, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Paiva, T.M.; Benega, M.A.; Silva, D.B.; Santos, K.C.; Cruz, A.S.; Hortenci, M.F.; Barbieri, M.T.; Monteiro, M.M.; Barbosa, H.A.; Carvalhanas, T.R. Evolutionary pattern of reemerging influenza B/Victoria-lineage viruses in São Paulo, Brazil, 1996–2012: Implications for vaccine composition strategy. J. Med. Virol. 2013, 85, 1983–1989. [Google Scholar] [CrossRef] [PubMed]
- Caini, S.; Huang, Q.S.; Ciblak, M.A.; Kusznierz, G.; Owen, R.; Wangchuk, S.; Henriques, C.M.; Njouom, R.; Fasce, R.A.; Yu, H.; et al. Global influenza B study, epidemiological and virological characteristics of influenza B: Results of the global influenza B study. Influenza Other Respir. Viruses 2015, 9, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansaldi, F.; D'Agaro, P.; de Florentiis, D.; Puzelli, S.; Lin, Y.P.; Gregory, V.; Bennett, M.; Donatelli, I.; Gasparini, R.; Crovari, P.; et al. Molecular characterization of influenza B viruses circulating in northern Italy during the 2001–2002 epidemic season. J. Med. Virol. 2003, 70, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Puzelli, S.; Frezza, F.; Fabiani, C.; Ansaldi, F.; Campitelli, L.; Lin, Y.P.; Gregory, V.; Bennett, M.; D'Agaro, P.; Campello, C.; et al. Changes in the hemagglutinins and neuraminidases of human influenza B viruses isolated in Italy during the 2001–2002, 2002–2003, and 2003–2004 seasons. J. Med. Virol. 2004, 74, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Skowronski, D.M.; Janjua, N.Z.; Sabaiduc, S.; de Serres, G.; Winter, A.L.; Gubbay, J.B.; Dickinson, J.A.; Fonseca, K.; Charest, H.; Bastien, N.; et al. Influenza A/subtype and B/lineage effectiveness estimates for the 2011–2012 trivalent vaccine: Cross-season and cross-lineage protection with unchanged vaccine. J. Infect. Dis. 2014, 210, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.C.; Chuang, J.H.; Kuo, H.W.; Huang, W.T.; Hsu, Y.F.; Liu, M.T.; Chen, C.H.; Huang, H.H.; Chang, C.H.; Chou, J.H.; et al. Surveillance and vaccine effectiveness of an influenza epidemic predominated by vaccine-mismatched influenza B/Yamagata-lineage viruses in Taiwan, 2011–2012 season. PLoS ONE 2013, 8, e58222. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, C.S.; Levin, M.J. The rationale for quadrivalent influenza vaccines. Hum. Vaccines Immunother. 2012, 8, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Ikonen, N.; Ziegler, T. Impact of influenza B lineage-level mismatch between trivalent seasonal influenza vaccines and circulating viruses, 1999–2012. Clin. Infect. Dis. 2014, 59, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Cheng, F.; Lu, M.; Tian, X.; Ma, J. Crystal structure of unliganded influenza B virus hemagglutinin. J. Virol. 2008, 82, 3011–3020. [Google Scholar] [CrossRef] [PubMed]
- Ni, F.; Kondrashkina, E.; Wang, Q. Structural basis for the divergent evolution of influenza B virus hemagglutinin. Virology 2013, 446, 112–122. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Influenza Virus Characterisation, Summary Europe. March 2013. Available online: http://ecdc.europa.eu/en/publications/Publications/influenza-virus-characterisation-mar-2013.pdf (accessed on 5 February 2016).
- European Centre for Disease Prevention and Control. Influenza Virus Characterisation, Summary Europe. June 2013. Available online: http://ecdc.europa.eu/en/publications/Publications/influenza-virus-characterisation-june-2013.pdf (accessed on 5 February 2016).
- European Centre for Disease Prevention and Control. Influenza Virus Characterisation, Summary Europe. June 2015. Available online: http://ecdc.europa.eu/en/publications/Publications/influenza-virus-characterisation-june-2015.pdf (accessed on 5 February 2016).
- Oong, X.Y.; Ng, K.T.; Lam, T.T.; Pang, Y.K.; Chan, K.G.; Hanafi, N.S.; Kamarulzaman, A.; Tee, K.K. Epidemiological and Evolutionary Dynamics of Influenza B Viruses in Malaysia, 2012–2014. PLoS ONE 2015, 10, e0136254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tewawong, N.; Suwannakarn, K.; Prachayangprecha, S.; Korkong, S.; Vichiwattana, P.; Vongpunsawad, S.; Poovorawan, Y. Molecular epidemiology and phylogenetic analyses of influenza B virus in Thailand during 2010 to 2014. PLoS ONE 2015, 10, e0116302. [Google Scholar]
- Chen, R.; Holmes, E.C. The evolutionary dynamics of human influenza B virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Influenza Surveillance Data. Available online: http://ecdc.europa.eu/en/healthtopics/seasonal_influenza/epidemiological_data/Pages/Latest_surveillance_data.aspx (accessed on 5 February 2016).
- Knossow, M.; Skehel, J.J. Variation and infectivity neutralization in influenza. Immunology 2006, 119, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Kirk, B.D.; Ma, J.; Wang, Q. Diversifying selective pressure on influenza B virus hemagglutinin. J. Med. Virol. 2009, 81, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Lin, Y.P.; Gregory, V.; Benett, M. Annual Report—WHO Influenza Centre; WHO: London, UK, 2001. [Google Scholar]
- Hay, A.J.; Lin, Y.P.; Gregory, V.; Benett, M. Annual Report—WHO Influenza Centre; WHO: London, UK, 2002. [Google Scholar]
- Ansaldi, F.; Bacilieri, S.; Amicizia, D.; Valle, L.; Banfi, F.; Durando, P.; Sticchi, L.; Gasparini, R.; Icardi, G.; Crovari, P. Antigenic characterisation of influenza B virus with a new microneutralisation assay: Comparison to haemagglutination and sequence analysis. J. Med. Virol. 2004, 74, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Ministero della Salute—Repubblica Italiana. InfluNet: Protocollo Operativo—Stagione Influenzale 2015–2016. Available online: http://www.salute.gov.it/imgs/C_17_pubblicazioni_2418_allegato.pdf (accessed on 5 February 2016).
- Tramuto, F.; Maida, C.M.; Bonura, F.; Perna, A.M.; Puzelli, S.; de Marco, M.A.; Donatelli, I.; Aprea, L.; Firenze, A.; Arcadipane, A.; et al. Surveillance of hospitalised patients with influenza-like illness during pandemic influenza A(H1N1) season in Sicily, April 2009–December 2010. Eur. Surveill. 2011, 16, 19957. [Google Scholar]
- World Health Organization. A revision of the system of nomenclature for influenza viruses: A WHO memorandum. Bull. World Health Organ. 1980, 58, 585–591. [Google Scholar]
- World Health Organization. WHO Information for Molecular Diagnosis of Influenza Virus. 27–29 March 2014. Available online: http://www.who.int/entity/influenza/gisrs_laboratory/molecular_diagnosis_influenza_virus_humans_update_201403rev201505.pdf?ua=1 (accessed on 5 February 2016).
- Global Initiative on Sharing All Influenza Data. Available online: http://platform.gisaid.org (accessed on 5 February 2016).
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html (accessed on 5 February 2016).
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subunit | Epitope (Residue Location) | B/Victoria | B/Yamagata | |
|---|---|---|---|---|
| Clade 1 (n = 29) | Clade 2 (n = 112) | Clade 3 (n = 56) | ||
| HA1 | 120-loop (116–137) | H116N (2) | ||
| T121S (1) | ||||
| H122Q (2) * | N116K (1) | N116K (47) | ||
| N126D (1) * | S120T (2) | Q122K (4) | ||
| N126S (1) ** | T121S (9) | N123T (4) | ||
| E128A (1) | D126N (1) | D126N (1) | ||
| N129K (2) | ||||
| N129D (4) | ||||
| 150-loop (141–150) | I146V (26) | T139A (1) | I150V (3) | |
| S150I (3) | I150S (1) | |||
| 160-loop (162–167) | N163D (1) | D163N (1) N165Y (3) | ||
| D164N (1) | ||||
| K165N (1) | - | |||
| K167R (1) | ||||
| K167N (1) | ||||
| 190-helix (194–202) | D197N (29) E198K (1) A202Q (1) A202K (1) | D196N (112) | D196N (54) | |
| K197E (1) | D196Y (1) | |||
| K201A (1) | K197R (3) * | |||
| N202K (1) | T198S (1) | |||
| N202S (2) | S202N (9) * | |||
| Influenza B Victoria-lineage Virus (Clade) | HI Titer Post Infection Ferret Antisera B/Brisbane/60/2008 F22/12 1A | Amino-Acid Mutations with Respect to B/Brisbane/60/2008 | |||
| 120-loop | 150-loop | ||||
| 37 | 58 | 80 | 146 | ||
| B/Brisbane/60/2008 (1A) | 1280 | V | L | R | I |
| B/Genoa/04/2010 (1A) | 320 | - | - | - | V |
| B/Genoa/08/2010 (1B) | 160 | - | P | - | V |
| B/Genoa/12/2011 (1B) | 160 | - | P | - | V |
| B/Genoa/22/2011 (1A) | 640 | I | - | G | - |
| Influenza B Yamagata-Lineage Virus (Clade) | HI Titer Post Infection Ferret Antisera | Amino-Acid Mutations with Respect to B/Wisconsin/01/2010 | |||||||||||||||||||||||||
| B/Wisc/ 01/10 | B/Mass/ 02/12 | B/Phuk/ 3073/13 | 120-loop | 150-loop | 160-loop | 190-helix | |||||||||||||||||||||
| 47 | 48 | 108 | 116 | 150 | 165 | 166 | 168 | 173 | 175 | 182 | 197 | 198 | 199 | 202 | 203 | 230 | 233 | 252 | 255 | 262 | 267 | 299 | 313 | ||||
| B/Wisc/01/10 (3C) | 1280 | 320 | 160 | T | R | P | N | I | N | Y | N | L | V | T | N | K | I | K | S | D | D | M | P | V | V | K | E |
| B/Mass/02/12 (2) | 1280 | 640 | 320 | - | K | A | - | S | - | N | - | - | - | A | - | - | T | - | N | - | - | - | - | - | - | - | - |
| B/Phuk/3073/13 (3) | 640 | 320 | 1280 | - | - | - | K | - | - | - | - | - | - | - | - | - | T | - | - | - | - | - | - | - | - | E | K |
| B/Genoa/0112/1213 (3) | 320 | 160 | 1280 | A | - | - | K | V | - | - | - | - | - | - | - | - | T | - | - | - | - | - | - | - | - | E | K |
| B/Genoa/0413/1213 (2) | 640 | 160 | 160 | - | K | A | - | S | - | N | - | - | - | A | - | - | T | - | N | G | - | - | - | - | - | - | - |
| B/Genoa/0613/1213 (3) | 160 | 160 | 640 | A | - | - | K | S | - | - | - | - | - | - | - | - | T | - | - | - | - | - | - | - | - | E | K |
| B/Genoa/0713/2013 (2) | 640 | 160 | 80 | - | K | A | - | S | - | N | - | - | - | A | - | - | T | - | N | G | - | - | - | - | - | - | - |
| B/Genoa/01/2014 (nd) | 160 | 80 | 80 | - | - | - | K | - | K | - | T | Q | I | - | - | E | T | A | K | G | N | V | S | T | I | - | - |
| B/Genoa/09/2015 (3) | 80 | 80 | 640 | - | - | - | K | - | - | - | - | - | - | - | D | - | T | - | - | - | - | - | - | - | - | E | K |
| B/Genoa/11/2015 (3) | 1280 | 320 | 2560 | - | - | - | K | - | - | - | - | Q | - | - | - | - | T | - | - | - | - | - | - | - | - | E | K |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tramuto, F.; Orsi, A.; Maida, C.M.; Costantino, C.; Trucchi, C.; Alicino, C.; Vitale, F.; Ansaldi, F. The Molecular Epidemiology and Evolutionary Dynamics of Influenza B Virus in Two Italian Regions during 2010–2015: The Experience of Sicily and Liguria. Int. J. Mol. Sci. 2016, 17, 549. https://doi.org/10.3390/ijms17040549
Tramuto F, Orsi A, Maida CM, Costantino C, Trucchi C, Alicino C, Vitale F, Ansaldi F. The Molecular Epidemiology and Evolutionary Dynamics of Influenza B Virus in Two Italian Regions during 2010–2015: The Experience of Sicily and Liguria. International Journal of Molecular Sciences. 2016; 17(4):549. https://doi.org/10.3390/ijms17040549
Chicago/Turabian StyleTramuto, Fabio, Andrea Orsi, Carmelo Massimo Maida, Claudio Costantino, Cecilia Trucchi, Cristiano Alicino, Francesco Vitale, and Filippo Ansaldi. 2016. "The Molecular Epidemiology and Evolutionary Dynamics of Influenza B Virus in Two Italian Regions during 2010–2015: The Experience of Sicily and Liguria" International Journal of Molecular Sciences 17, no. 4: 549. https://doi.org/10.3390/ijms17040549
APA StyleTramuto, F., Orsi, A., Maida, C. M., Costantino, C., Trucchi, C., Alicino, C., Vitale, F., & Ansaldi, F. (2016). The Molecular Epidemiology and Evolutionary Dynamics of Influenza B Virus in Two Italian Regions during 2010–2015: The Experience of Sicily and Liguria. International Journal of Molecular Sciences, 17(4), 549. https://doi.org/10.3390/ijms17040549