
Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease



 ,
,
Abstract
:1. Introduction
2. Results
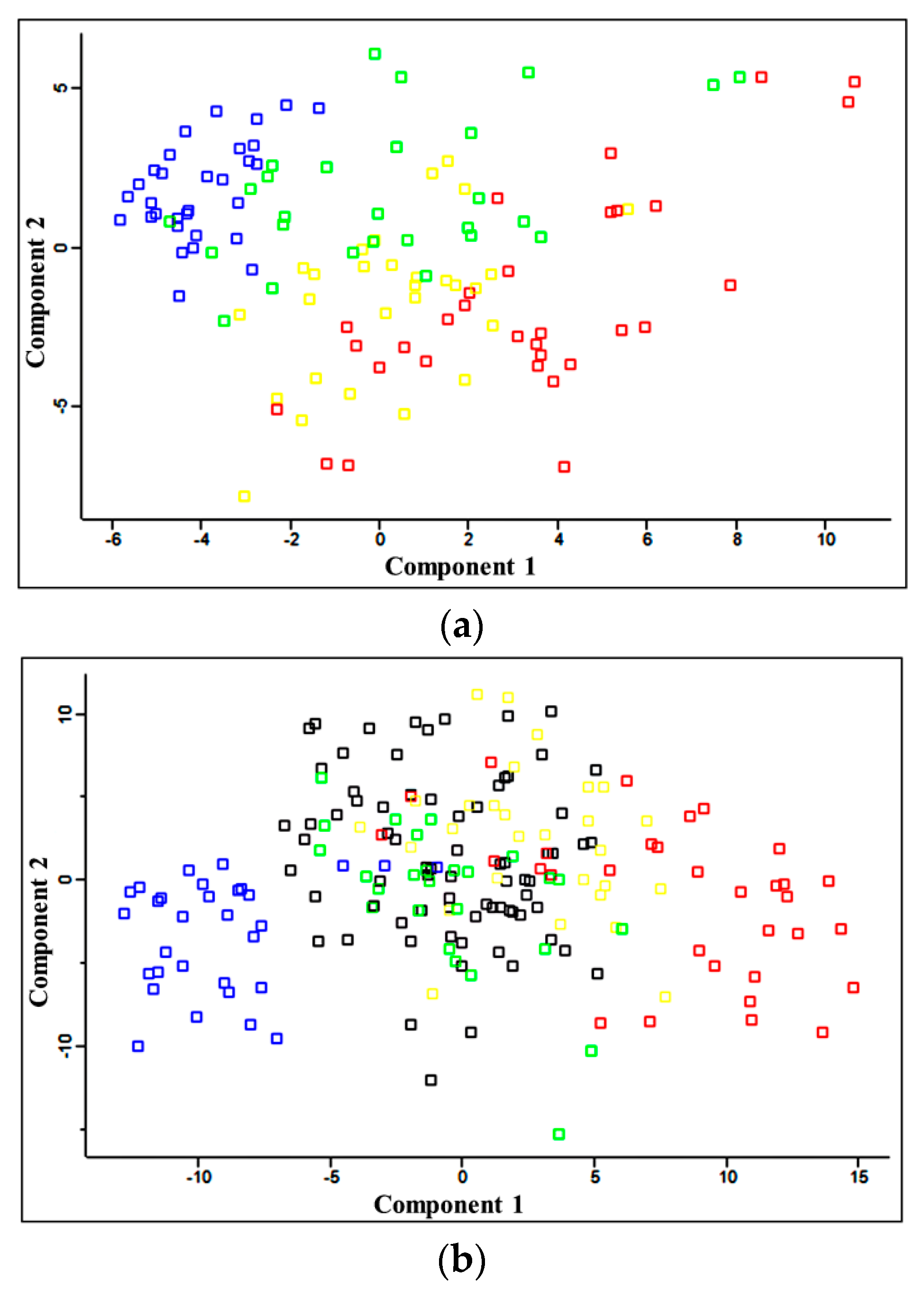
2.1. Quantitative Analysis of Plasma Proteins
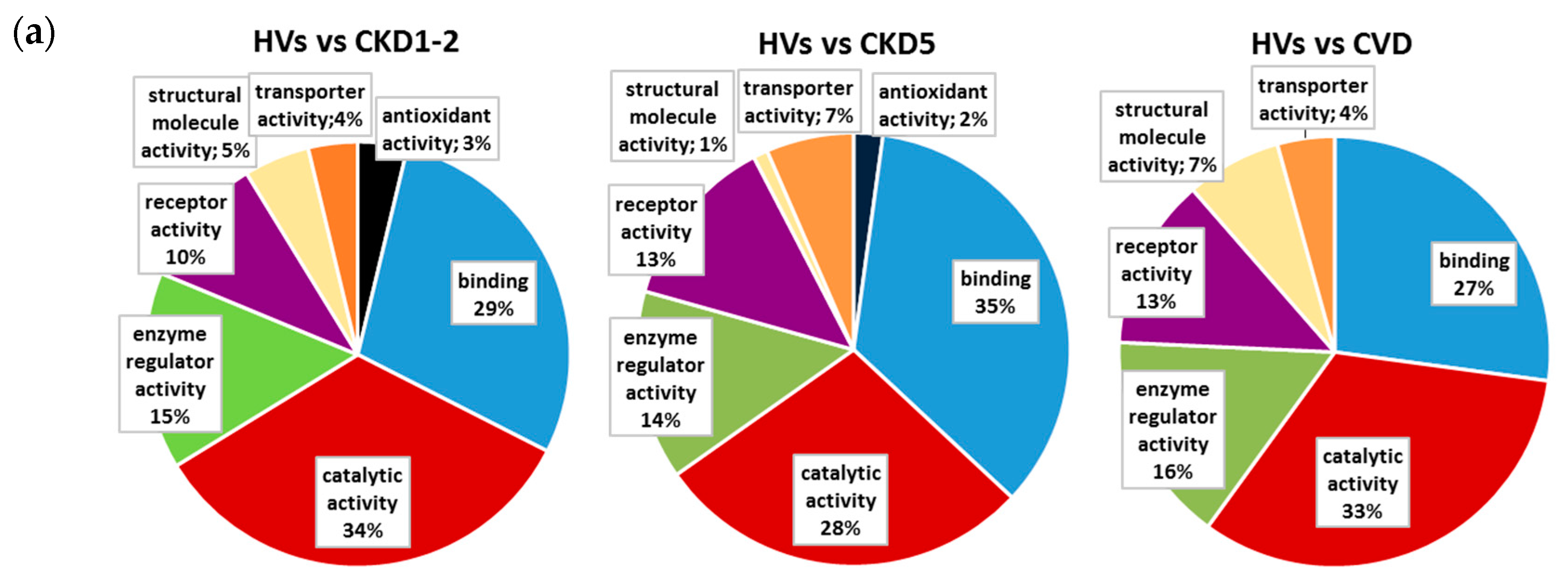
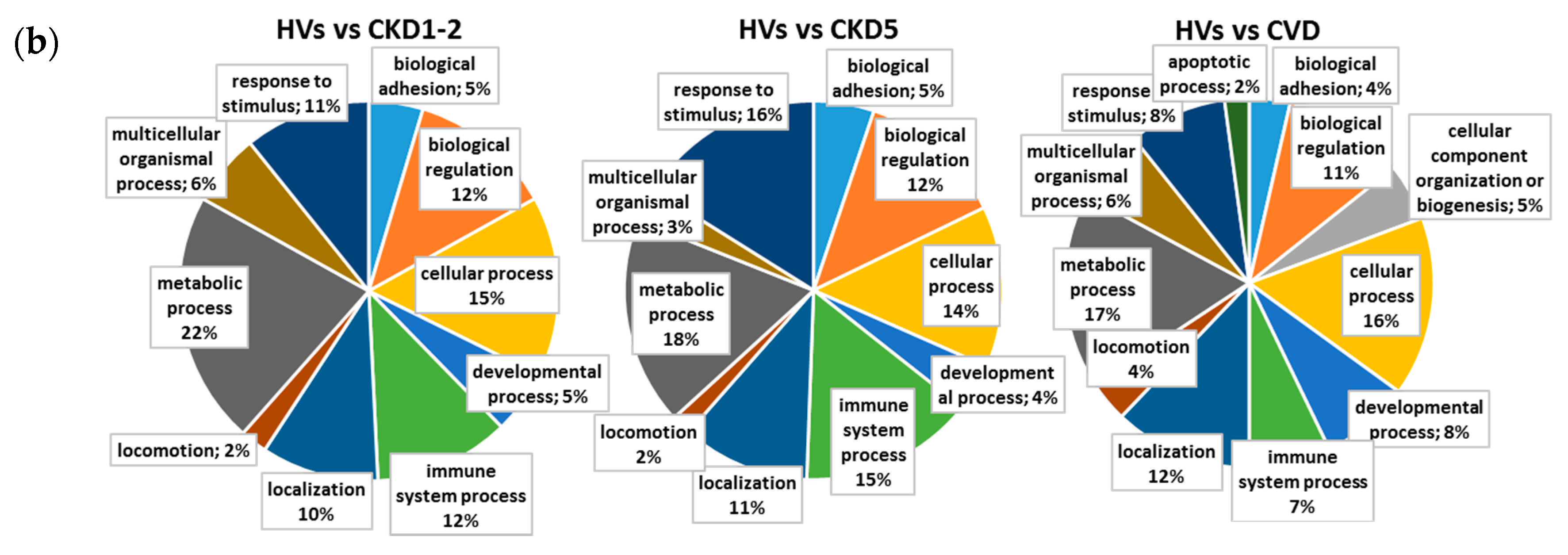
2.2. Pathways and Functional Annotations of Differential Proteins
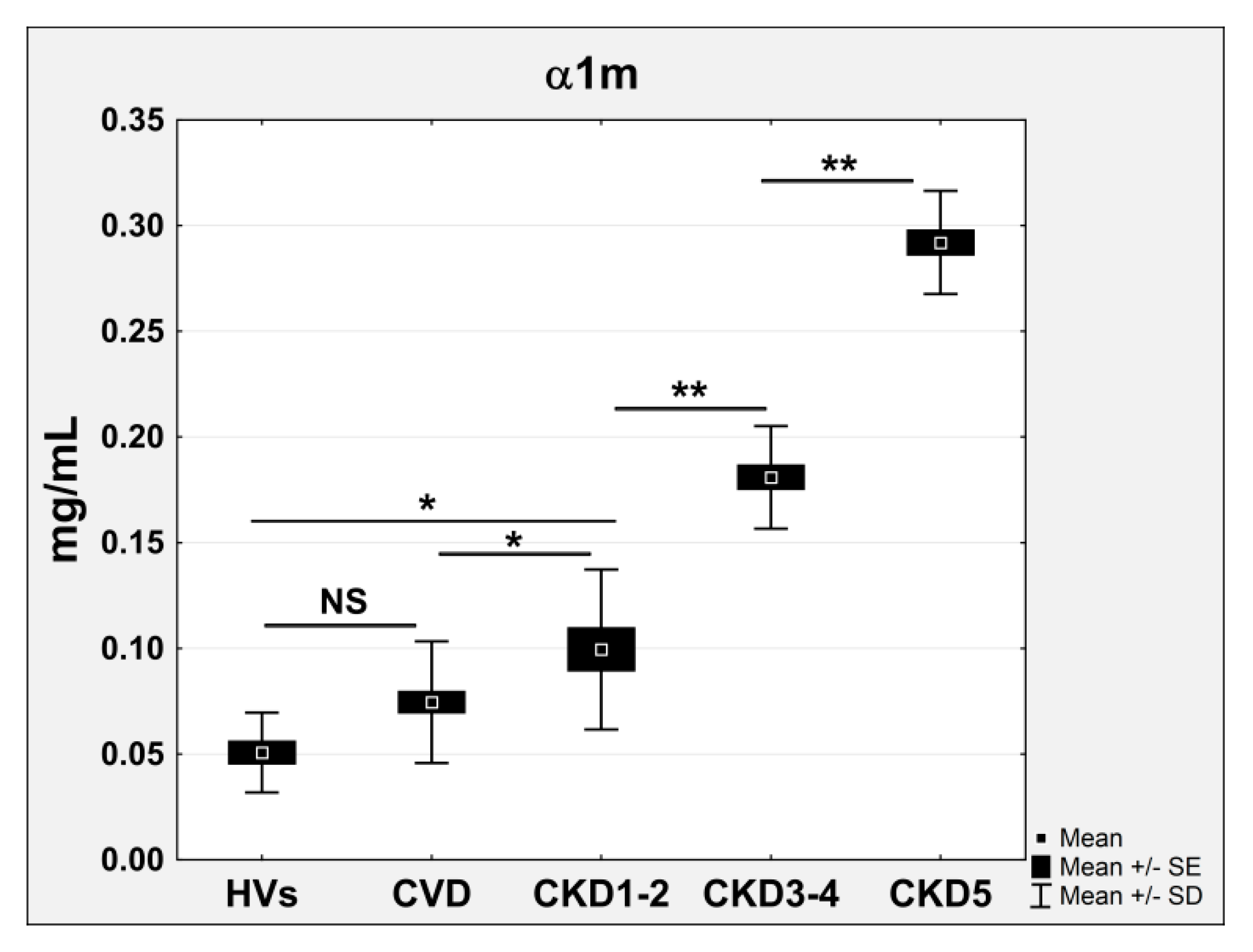
2.3. Proteins Specifically Related to CKD Progression
3. Discussion
4. Materials and Methods
4.1. Subjects and Samples
4.2. In-Solution Trypsin Digestion
4.3. NanoLC-MS/MS Analysis
4.4. Qualitative Analysis of Proteomic Data
4.5. Quantitative Analysis of Proteomic Data
4.6. Assessment of Variability/Reproducibility
4.7. ELISA Validation
4.8. Pathway and Network Analyses of Dysregulated Proteins in Plasma Samples
4.9. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CKD | chronic kidney disease |
| CVD | cardiovascular disease |
| CKD-A | CKD-related atherosclerosis |
| HV | healthy volunteer |
| GFR | glomerular filtration rate |
| LFQ | label-free quantification |
| LC-MS/MS | liquid chromatography tandem mass spectrometry |
| DTT | dithiothreitol |
| α1m | α-1-microglobulin |
| β2m | β-2-microglobulin |
| cysC | cystatin C |
References
- Liabeuf, S.; Desjardins, L.; Diouf, M.; Temmar, M.; Renard, C.; Choukroun, G.; Massy, Z.A. The addition of vascular calcification scores to traditional risk factors improves cardiovascular risk assessment in patients with chronic kidney disease. PLoS ONE 2015, 10, e0131707. [Google Scholar] [CrossRef] [PubMed]
- Said, A.; Desai, C.; Lerma, E.V. Chronic kidney disease. Dis. Mon. 2015, 61, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Reriani, M.K.; Lerman, L.O.; Lerman, A. Endothelial function as a functional expression of cardiovascular risk factors. Biomark. Med. 2010, 4, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.M.; Pinto, D.S.; Murphy, S.A.; Morrow, D.A.; Hobbach, H.P.; Wiviott, S.D.; Giugliano, R.P.; Cannon, C.P.; Antman, E.M.; Braunwald, E. Association of creatinine and creatinine clearance on presentation in acute myocardial infarction with subsequent mortality. J. Am. Coll. Cardiol. 2003, 42, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Lill, J. Proteomic tools for quantitation by mass spectrometry. Mass Spectrom. Rev. 2003, 22, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass spectrometry-based proteomics. Nature 2003, 422, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Fuller, H.; Morris, G. Quantitative proteomics using iTRAQ labeling and mass spectrometry. Integr. Proteom. 2012, 347–342. [Google Scholar]
- Luczak, M.; Marczak, L.; Stobiecki, M. Optimization of plasma sample pretreatment for quantitative analysis using iTRAQ labeling and LC-MALDI-TOF/TOF. PLoS ONE 2014, 9, e101694. [Google Scholar] [CrossRef] [PubMed]
- Schindler, R. Causes and therapy of microinflammation in renal failure. Nephrol. Dial. Transplant. 2004, 19, V34–V40. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Lucchi, L.; Bergamini, S.; Iannone, A.; Perrone, S.; Stipo, L.; Olmeda, F.; Caruso, F.; Tomasi, A.; Albertazzi, A. Erythrocyte susceptibility to oxidative stress in chronic renal failure patients under different substitutive treatments. Artif. Organs 2005, 29, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.-H.; Wang, C.-L.; Chen, P.-C.; Yang, T.-C. Linkage of some trace elements, peripheral blood lymphocytes, inflammation, and oxidative stress in patients undergoing either hemodialysis or peritoneal dialysis. Perit. Dial. Int. 2011, 31, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Salgado, J.V.; França, A.K.; Cabral, N.A.; Lages, J.; Ribeiro, V.S.; Santos, A.M.; Salgado, B.J. Cystatin C, kidney function, and cardiovascular risk factors in primary hypertension. Rev. Assoc. Méd. Bras. 2013, 59, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Dharnidharka, V.R.; Kwon, C.; Stevens, G. Serum cystatin C is superior to serum creatinine as a marker of kidney function: A meta-analysis. Am. J. Kidney Dis. 2002, 40, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Filler, G.; Bökenkamp, A.; Hofmann, W.; Le Bricon, T.; Martínez-Brú, C.; Grubb, A. Cystatin C as a marker of GFR—History, indications, and future research. Clin. Biochem. 2005, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yılmaz, B. Serum β2-microglobulin as a biomarker in inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 10916. [Google Scholar] [CrossRef] [PubMed]
- Raikou, V.D.; Kyriaki, D. The relationship between glycemic control, β2-microglobulin and inflammation in patients on maintenance dialysis treatment. J. Diabetes Metab. Disord. 2015, 14. [Google Scholar] [CrossRef] [PubMed]
- Salgado, J.V.; Neves, F.A.; Bastos, M.G.; França, A.K.; Brito, D.J.; Santos, E.M.; Salgado Filho, N. Monitoring renal function: Measured and estimated glomerular filtration rates—A review. Braz. J. Med. Biol. Res 2010, 43, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Batra, A.; Kapoor, A.; Sharma, R.K.; Agrawal, N.; Sinha, A.; Kumar, S.; Garg, N.; Tewari, S.; Goel, P.K. Association of plasma cystatin C levels with angiographically documented coronary artery disease in patients of Indian origin. J. Cardiol. 2012, 59, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Loew, M.; Hoffmann, M.M.; Koenig, W.; Brenner, H.; Rothenbacher, D. Genotype and plasma concentration of cystatin C in patients with coronary heart disease and risk for secondary cardiovascular events. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Formanowicz, D.; Marczak, Ł.; Pawliczak, E.; Wanic-Kossowska, M.; Figlerowicz, M.; Stobiecki, M. Deeper insight into chronic kidney disease-related atherosclerosis: Comparative proteomic studies of blood plasma using 2DE and mass spectrometry. J. Transl. Med. 2015, 13. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Pecoits-Filho, R. Vascular damage in kidney disease: Beyond hypertension. Int. J. Hypertens. 2011, 2011, 232683. [Google Scholar] [CrossRef] [PubMed]
- Pickering, W.P.; Price, S.R.; Bircher, G.; Marinovic, A.C.; Mitch, W.E.; Walls, J. Nutrition in CAPD: Serum bicarbonate and the ubiquitin-proteasome system in muscle. Kidney Int. 2002, 61, 1286–1292. [Google Scholar] [CrossRef] [PubMed]
- Zachara, B.A.; Gromadzińska, J.; Wasowicz, W.; Zbróg, Z. Red blood cell and plasma glutathione peroxidase activities and selenium concentration in patients with chronic kidney disease: A review. Acta Biochim. Pol. 2006, 53, 663–677. [Google Scholar] [PubMed]
- Goodman, W.G.; London, G.; Amann, K.; Block, G.A.; Giachelli, C.; Hruska, K.A.; Ketteler, M.; Levin, A.; Massy, Z.; McCarron, D.A.; et al. Vascular calcification in chronic kidney disease. Am. J. Kidney Dis. 2004, 43, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Mazière, C.; Kamel, S.; Brazier, M.; Choukroun, G.; Tribouilloy, C.; Slama, M.; Andrejak, M.; Mazière, J.C. Impact of inflammation and oxidative stress on vascular calcifications in chronic kidney disease. Pediatr. Nephrol. 2005, 20, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Wade, A.N.; Reilly, M.P. Coronary calcification in chronic kidney disease: Morphology, mechanisms and mortality. Clin. J. Am. Soc. Nephrol. 2009, 4, 1883–1885. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Chen, Y. Molecular mechanisms of vascular calcification in chronic kidney disease: The link between bone and the vasculature. Curr. Osteoporos. Rep. 2015, 13, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Berezin, A.E.; Kremzer, A.A. Circulating osteopontin as a marker of early coronary vascular calcification in type two diabetes mellitus patients with known asymptomatic coronary artery disease. Atherosclerosis 2013, 229, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Siasos, G.; Maniatis, K.; Oikonomou, E.; Kioufis, S.; Zaromitidou, M.; Paraskevopoulos, T.; Michalea, S.; Kollia, C.; Miliou, A.; et al. Serum osteoprotegerin and osteopontin levels are associated with arterial stiffness and the presence and severity of coronary artery disease. Int. J. Cardiol. 2013, 167, 1924–1928. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, J.; Krämer, R.; Kliem, V.; Bode-Boeger, S.M.; Veldink, H.; Haller, H.; Fliser, D.; Kielstein, J.T. Circulating levels of osteopontin are closely related to glomerular filtration rate and cardiovascular risk markers in patients with chronic kidney disease. Eur. J. Clin. Investig. 2010, 40, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Heiss, A.; Eckert, T.; Aretz, A.; Richtering, W.; van Dorp, W.; Schäfer, C.; Jahnen-Dechent, W. Hierarchical role of fetuin-A and acidic serum proteins in the formation and stabilization of calcium phosphate particles. J. Biol. Chem. 2008, 283, 14815–14825. [Google Scholar] [CrossRef] [PubMed]
- Jahnen-Dechent, W.; Heiss, A.; Schäfer, C.; Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef] [PubMed]
- Denecke, B.; Gräber, S.; Schäfer, C.; Heiss, A.; Wöltje, M.; Jahnen-Dechent, W. Tissue distribution and activity testing suggest a similar but not identical function of fetuin-B and fetuin-A. Biochem. J. 2003, 376, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; de Las Fuentes, L.; Bierhals, A.; Ash-Bernal, R.; Spence, K.; Slatopolsky, E.; Davila-Roman, V.G.; Delmez, J. Relation of serum fetuin-A levels to coronary artery calcium in African-American patients on chronic hemodialysis. Am. J. Cardiol. 2009, 103, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Emoto, M.; Mori, K.; Lee, E.; Kawano, N.; Yamazaki, Y.; Tsuchikura, S.; Morioka, T.; Koyama, H.; Shoji, T.; Inaba, M.; et al. Fetuin-A and atherosclerotic calcified plaque in patients with type 2 diabetes mellitus. Metabolism 2010, 59, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 16, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; ten Cate, H.; van der Poll, T. Endothelium: Interface between coagulation and inflammation. Crit. Care Med. 2002, 30, S220–S224. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Formanowicz, D.; Pawliczak, E.; Wanic-Kossowska, M.; Wykretowicz, A.; Figlerowicz, M. Chronic kidney disease-related atherosclerosis—Proteomic studies of blood plasma. Proteome Sci. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Minnema, M.C.; Peters, R.J.; de Winter, R.; Lubbers, Y.P.; Barzegar, S.; Bauer, K.A.; Rosenberg, R.D.; Hack, C.E.; ten Cate, H. Activation of clotting factors XI and IX in patients with acute myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2489–2493. [Google Scholar] [CrossRef] [PubMed]
- Khrenov, A.V.; Ananyeva, N.M.; Griffin, J.H.; Saenko, E.L. Coagulation pathways in atherothrombosis. Trends Cardiovasc. Med. 2002, 12, 317–324. [Google Scholar] [CrossRef]
- Shlipak, M.G.; Fried, L.F.; Crump, C.; Bleyer, A.J.; Manolio, T.A.; Tracy, R.P.; Furberg, C.D.; Psaty, B.M. Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation 2003, 107, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Stevens, P.; Bilous, R. Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, e150. [Google Scholar]
- Chronic Kidney Disease (Partial Update): Early Identification and Management of Chronic Kidney Disease in Adults in Primary and Secondary Care; NICE Clinical Guidelines, No. 182; National Clinical Guideline Centre: London, UK, 2014; pp. 113–120.
- Levey, A.S.; Bosch, J.P.; Lewis, J.B.; Greene, T.; Rogers, N.; Roth, D. A more accurate method to estimate glomerular filtration rate from serum creatinine: A new prediction equation. Modification of Diet in Renal Disease Study Group. Ann. Intern. Med. 1999, 130, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Poudel, S.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2015, 44, D336–D342. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Group | Correlation Coefficients in Biological Replicates | Correlation Coefficients in Technical Replicates |
|---|---|---|
| HVs | 0.9103–0.9887 | 0.9894–0.9967 |
| CKD1-2 | 0.8711–0.9747 | 0.9784–0.9960 |
| CKD3-4 | 0.8603–0.9774 | 0.9531–0.9923 |
| CKD5 | 0.8391–0.9757 | 0.9196–0.9857 |
| CVD | 0.8110–0.9721 | 0.9466–0.9975 |
| Pathway | Database | HV/CKD1-2 | HV/CKD5 | HV/CVD | Benjamini Corrected p-Value |
|---|---|---|---|---|---|
| (% of Whole Proteins) | (% of Whole Proteins) | (% of Whole Proteins) | |||
| Hemostasis | REACTOME | 23.8 | 19.7 | 23.3 | 3.1 × 10−6/4.2 × 10−7/9.6 × 10−6 |
| Complement cascade | KEGG | 23.9 | 13.6 | 9.3 | 3.6 × 10−11/4.1 × 10−8/4.5 × 10−2 |
| Blood coagulation | PANTHER | 17.5 | 25 | 13.7 | 1.2 × 10−1/5.7 × 10−12/1.8 × 10−7 |
| Inflammation mediated by chemokine and cytokine signaling pathway | PANTHER | 8.3 | 8.3 | 4.8 | 2.5 × 10−5/6.4 × 10−5/2.4 × 10−6 |
| Integrin cell surface interaction | REACTOME | 15.2 | 12.1 | – | 1.1 × 10−4/1.5 × 10−6/NS |
| Signaling in immune system | REACTOME | 12.1 | 19.6 | – | 2.8 × 10−3/2.5 × 10−3/NS |
| Plasminogen activation cascade | PANTHER | 7.6 | 10.9 | – | 5.6 × 10−5/7.1 × 10−5/NS |
| Cardiac muscle contraction | KEGG | – | – | 9.3 | NS/NS/3.2 × 10−2 |
| Cardiomyopathy | KEGG | – | – | 14.6 | NS/NS/2.7 × 10−2 |
| Metabolism of lipids and lipoproteins | PANTHER | 8.3 | 3.1 | 8.3 | 2.4 × 10−4/2.1 × 10−3/2.8 × 10−5 |
| Protein | Correlation Coefficient | ANOVA | CKD1-2/HV | CKD3-4/HV | CKD5/HV | CVD/HV | Pathway/Process |
|---|---|---|---|---|---|---|---|
| Transferrin | 0.750 | 8.6 × 10−11 | 0.88 | 0.62 | 0.56 | 0.94 | Hemostasis |
| Vitronectin | 0.770 | 1.7 × 10−17 | 0.91 | 0.77 | 0.65 | 0.99 | Hemostasis |
| Hepatocyte growth factor activator | 0.719 | 0.0041 | 0.77 | 0.47 | 0.53 | 0.71 | Hemostasis |
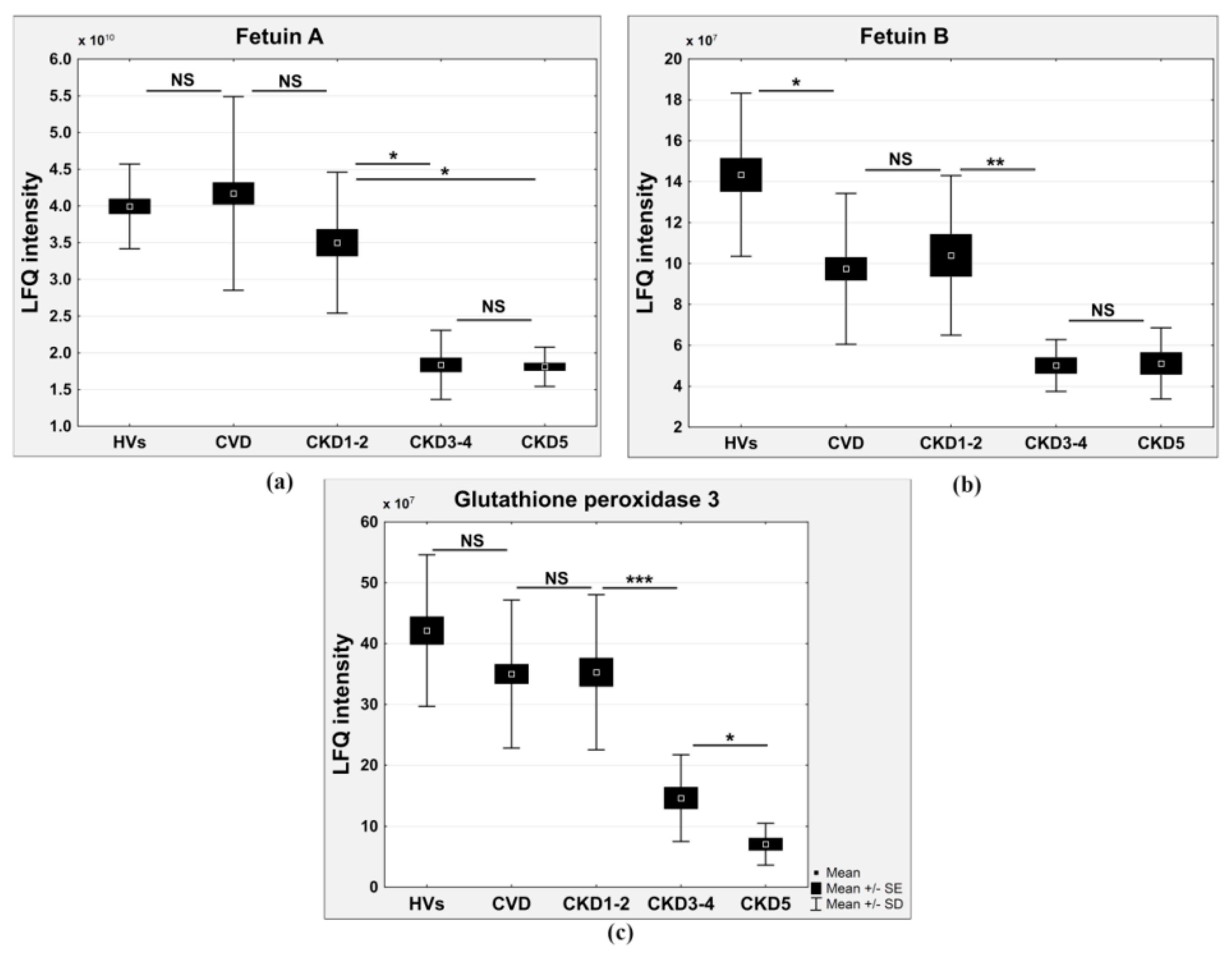
| Glutathione peroxidase 3 | 0.760 | 3 × 10−14 | 0.81 | 0.33 | 0.16 | 0.8 | Reactive oxygen species (ROS) detoxification |
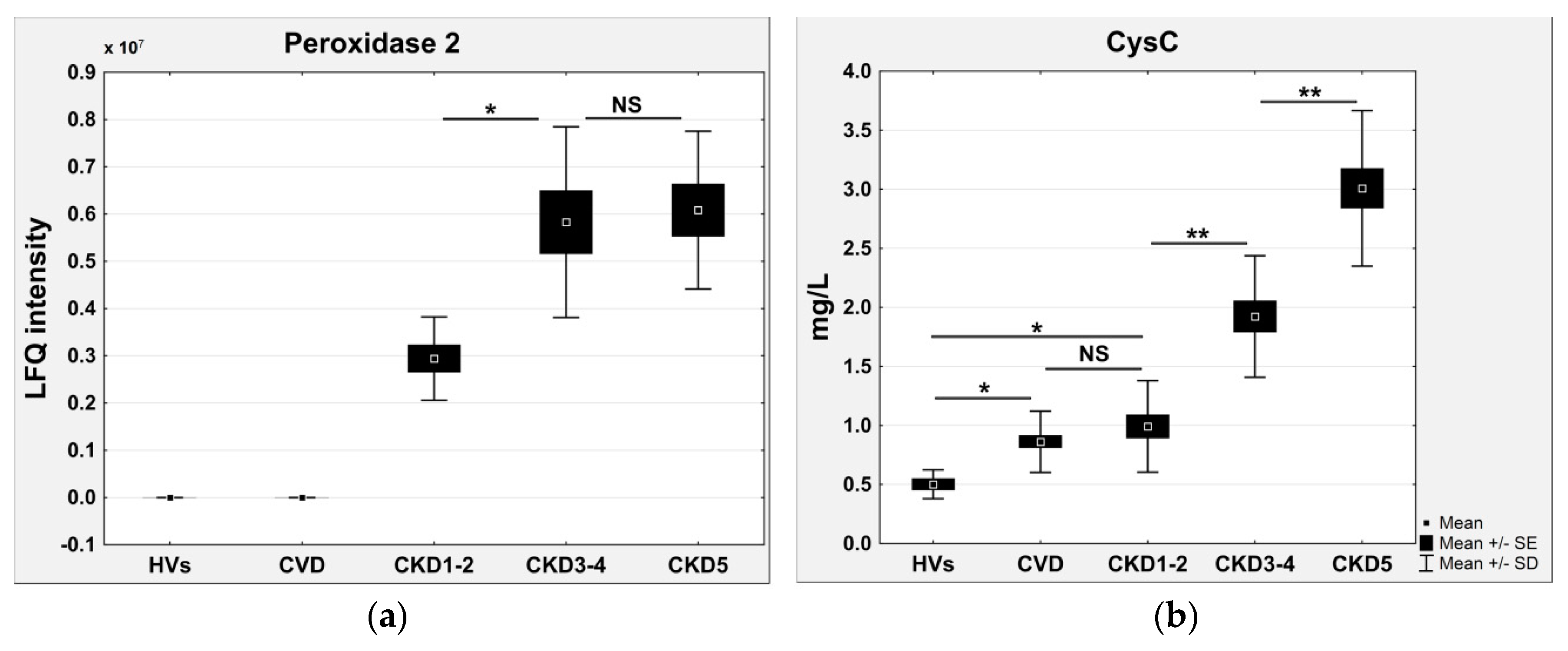
| Peroxiredoxin-2 | −0.7195 | 0.0049 | – | 2.03 | 2.06 | – | ROS detoxification |
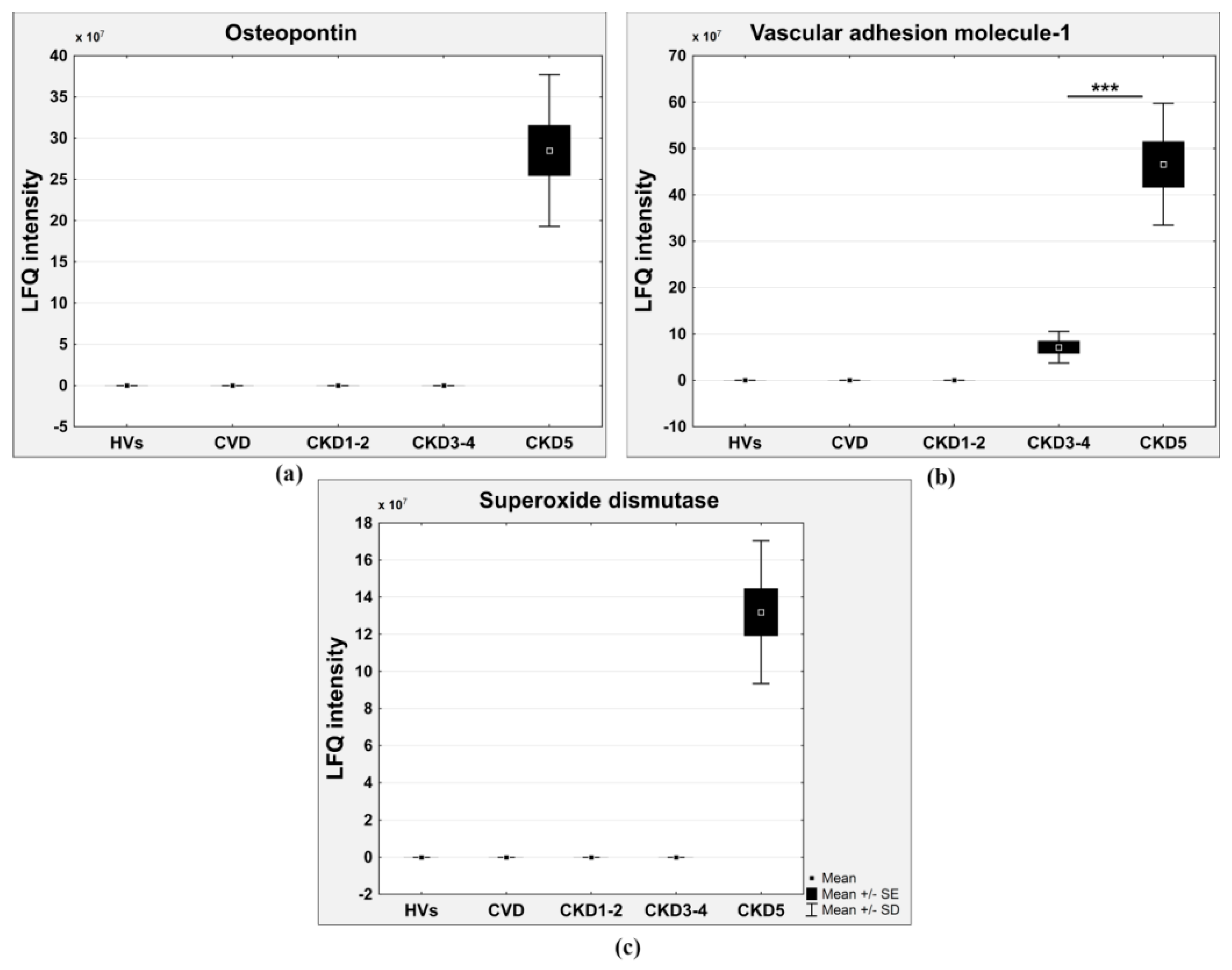
| Superoxide dismutase | present only in CKD5 | 0.0243 | – | – | – | – | ROS detoxification |
| Fetuin A | 0.730 | 0.0451 | 0.87 | 0.5 | 0.46 | 1.04 | Calcium metabolism |
| Fetuin-B | 0.779 | 6.1 × 10−5 | 0.71 | 0.35 | 0.4 | 0.69 | Calcium metabolism |
| Fibrinogen α | −0.735 | 1.5 × 10−13 | 1.59 | 1.76 | 1.85 | 1.34 | Complement and hemostasis |
| Fibrinogen β | −0.770 | 1.2 × 10−12 | 1.59 | 1.85 | 2.05 | 1.45 | Complement and hemostasis |
| Fibrinogen γ | −0.735 | 3.9 × 10−11 | 1.61 | 1.63 | 1.9 | 1.19 | Complement and hemostasis |
| β2m | −0.791 | 2.2 × 10−44 | 2.46 | 8.09 | 32.04 | 1.52 | Signaling in immune system |
| Complement component C7 | −0.797 | 0.0013 | 1.04 | 1.25 | 1.75 | 1.02 | Complement and blood coagulation, immune response |
| Complement factor H-related protein 1 | −0.706 | 5.5 × 10−13 | 1.49 | 1.43 | 2.05 | 1.17 | Complement and blood coagulation, immune response |
| Coagulation factor XIII B chain | −0.720 | 2.4 × 10−18 | 1.2 | 1.22 | 1.66 | 1.04 | Complement and blood coagulation, immune response |
| EGF-containing fibulin-like extracellular matrix protein 1 | −0.740 | 8.8 × 10−13 | 1.7 | 2.47 | 2.45 | 0.88 | Molecules associated with elastic fibers |
| Inter-α-trypsin inhibitor heavy chain H3 | −0.732 | 7 × 10−9 | 1.08 | 1.57 | 1.63 | 1.4 | No hits |
| Leucine-rich α-2-glycoprotein | −0.701 | 3.4 × 10−9 | 1.06 | 1.82 | 2.04 | 1.57 | No hits |
| Peptidase inhibitor 16 | −0.681 | 4.6 × 10−15 | 1.09 | 2.89 | 3.57 | 1.03 | No hits |
| Guanylin | present only in CKD5 | 8.2 × 10−13 | – | – | – | – | No hits |
| Protein AMBP; α1m | −0.790 | 5.1 × 10−54 | 2.04 | 2.86 | 4.89 | 1.4 | Scavenging of heme from plasma, inflammation mediated by chemokine and cytokine signaling |
| Apolipoprotein C-III | −0.761 | 0.0003 | 1.33 | 1.58 | 1.61 | 1.02 | Metabolism of lipids and lipoproteins |
| α-1-acid glycoprotein2 | −0.706 | 2.2 × 10−6 | 1.26 | 1.27 | 1.52 | 1.11 | Regulation and signaling in immune system |
| α-1-acid glycoprotein1 | −0.749 | 3 × 10−8 | 1.32 | 1.47 | 1.72 | 1.27 | Regulation and signaling in immune system |
| Retinol-binding protein 4 | −0.770 | 7.3 × 10−38 | 1.35 | 1.94 | 3.29 | 0.91 | Retinoid metabolism and transport |
| CysC | −0.826 | 8.4 × 10−27 | – | 4.25 | 6.32 | 0.85 | Response to stimuli, cellular response to oxidative stress |
| Zinc-α-2glycoprotein | −0.716 | 2.6 × 10−22 | 1.1 | 1.86 | 2.27 | 1.39 | Immune response, miscellaneous transport and binding events |
| Lumican | −0.769 | 2.7 × 10−13 | 1.07 | 1.38 | 1.58 | 1.06 | Integrin cell surface interactions |
| β-2-glycoprotein 1 | −0.813 | 1.4 × 10−7 | 1.2 | 1.54 | 1.63 | 1.2 | Blood coagulation |
| Pigment epithelium-derived factor | −0.799 | 9.7 × 10−45 | 1.26 | 1.61 | 2.12 | 1.11 | Blood coagulation |
| Monocyte differentiation antigen CD14 | −0.771 | 0.0001 | 2.02 | 2.85 | 3.68 | 1.55 | Immune response |
| Vascular cell adhesion molecule 1 | present only in CKD3-4 and CKD5 | 0.0312 | – | – | – | – | Integrin cell surface interactions, immune response |
| Prostaglandin-H2 d-isomerase | present only in CKD3-4 and CKD5 | 7.4 × 10−2 | – | – | – | – | Synthesis of prostaglandins and thromboxanes, hemostasis |
| Osteopontin | present only in CKD5 | 4 × 10−1 | – | – | – | – | Integrin cell surface interactions |
| Calreticulin | present only in CKD5 | 0.0479 | – | – | – | – | Calcium ion binding, chaperone |
| CD59 glycoprotein | present only in CKD5 | 5.9 × 10−11 | – | – | – | – | Regulation of complement cascade |
| Uteroglobin | present only in CKD5 | 1.6 × 10−1 | – | – | – | – | Immune response |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luczak, M.; Suszynska-Zajczyk, J.; Marczak, L.; Formanowicz, D.; Pawliczak, E.; Wanic-Kossowska, M.; Stobiecki, M. Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease. Int. J. Mol. Sci. 2016, 17, 631. https://doi.org/10.3390/ijms17050631
Luczak M, Suszynska-Zajczyk J, Marczak L, Formanowicz D, Pawliczak E, Wanic-Kossowska M, Stobiecki M. Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease. International Journal of Molecular Sciences. 2016; 17(5):631. https://doi.org/10.3390/ijms17050631
Chicago/Turabian StyleLuczak, Magdalena, Joanna Suszynska-Zajczyk, Lukasz Marczak, Dorota Formanowicz, Elzbieta Pawliczak, Maria Wanic-Kossowska, and Maciej Stobiecki. 2016. "Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease" International Journal of Molecular Sciences 17, no. 5: 631. https://doi.org/10.3390/ijms17050631
APA StyleLuczak, M., Suszynska-Zajczyk, J., Marczak, L., Formanowicz, D., Pawliczak, E., Wanic-Kossowska, M., & Stobiecki, M. (2016). Label-Free Quantitative Proteomics Reveals Differences in Molecular Mechanism of Atherosclerosis Related and Non-Related to Chronic Kidney Disease. International Journal of Molecular Sciences, 17(5), 631. https://doi.org/10.3390/ijms17050631