
Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects

 ,
, 
Abstract
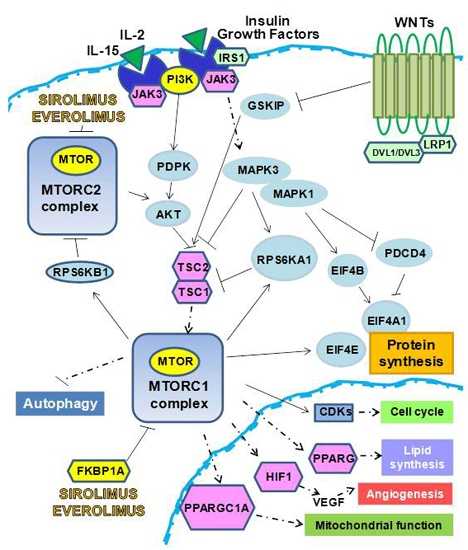
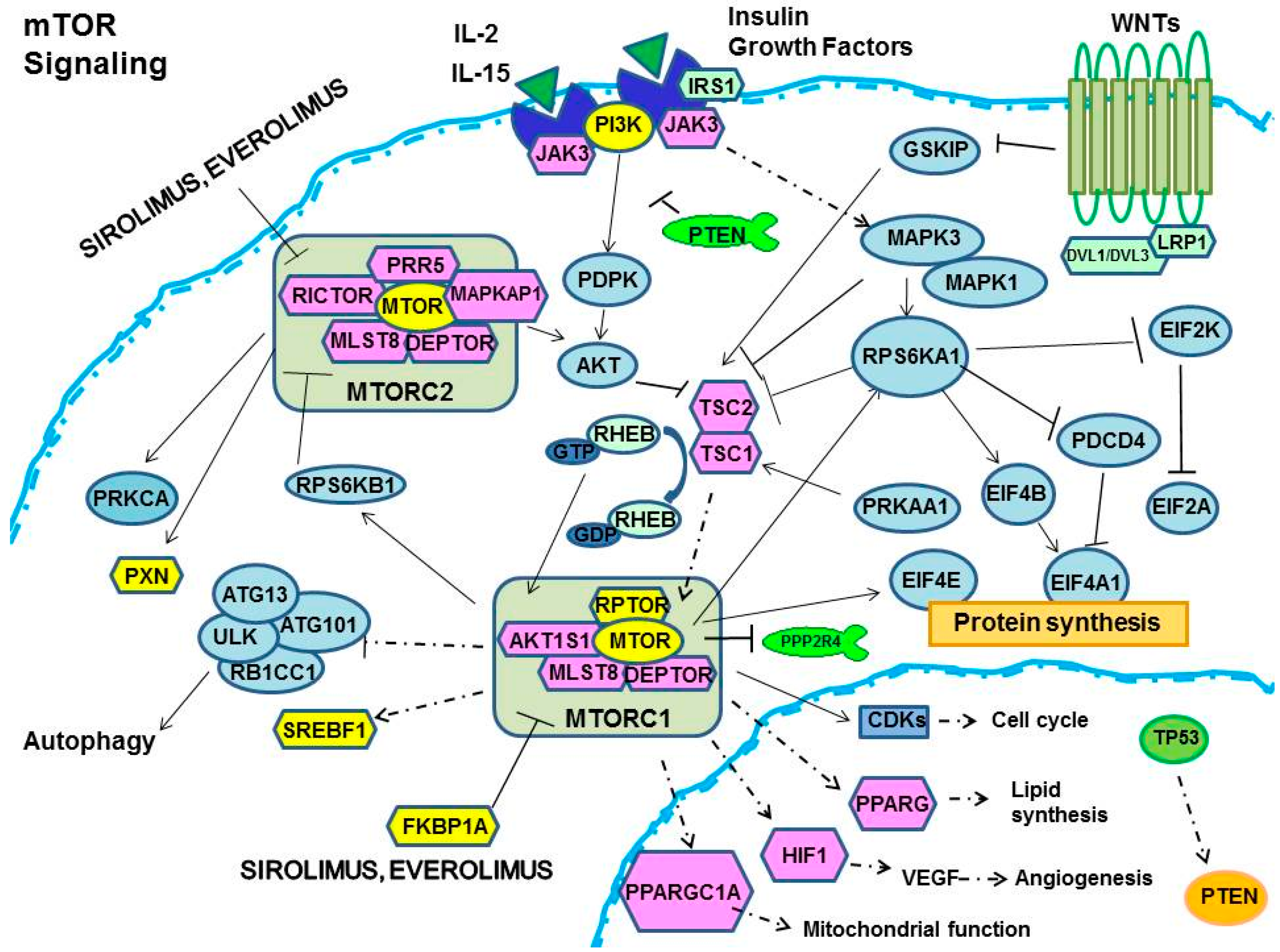
: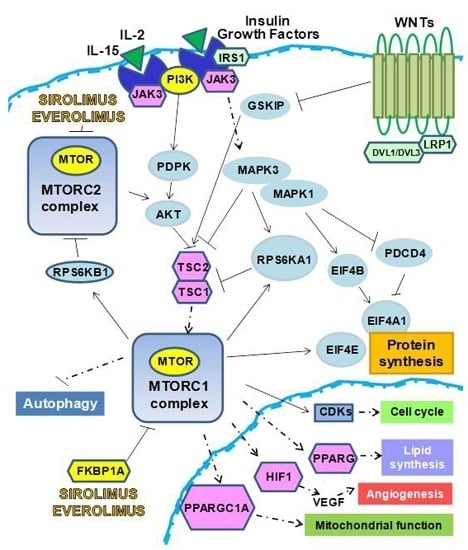
1. m-TOR Inhibitors (mTOR-I): Clinical Aspects
2. The Biological Effects of mTOR-I
2.1. Control of Protein Synthesis by m-TOR-I
2.2. mTOR-I Regulation of Angiogenesis
2.3. mTOR Inhibition and Lipid Biosynthesis
2.4. Biochemical mTOR-I-Related Mitochondrial Biogenesis and Functional Regulation
2.5. m-TOR-Is Control Cell Cycle and Growth
2.6. mTOR-I and Autophagy
3. MicroRNA (miRNAs) and mTOR
4. Pharmacogenetics/Genomics and mTOR-I
Author Contributions
Conflicts of Interest
References
- Kirchner, G.I.; Meier-Wiedenbach, I.; Manns, M.P. Clinical pharmacokinetics of everolimus. Clin. Pharmacokinet. 2004, 43, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, S.N. Rapamune (RAPA, rapamycin, sirolimus): Mechanism of action immunosuppressive effect results from blockade of signal transduction and inhibition of cell cycle progression. Clin. Biochem. 1998, 31, 335–340. [Google Scholar] [CrossRef]
- Kelly, P.A.; Gruber, S.A.; Behbod, F.; Kahan, B.D. Sirolimus, a new, potent immunosuppressive agent. Pharmacotherapy 1997, 17, 1148–1156. [Google Scholar] [PubMed]
- Morath, C.; Arns, W.; Schwenger, V.; Mehrabi, A.; Fonouni, H.; Schmidt, J.; Zeier, M. Sirolimus in renal transplantation. Nephrol. Dial. Transplant. 2007, 22 (Suppl. 8), viii61–viii65. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, E.; Ratto, E.; Bellino, D.; Marsano, L.; Cassottana, P.; Cannella, G. Effect of early conversion from CNI to sirolimus on outcomes in kidney transplant recipients with allograft dysfunction. J. Nephrol. 2012, 25, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Schena, F.P.; Pascoe, M.D.; Alberu, J.; del Carmen Rial, M.; Oberbauer, R.; Brennan, D.C.; Campistol, J.M.; Racusen, L.; Polinsky, M.S.; Goldberg-Alberts, R.; et al. Conversion from calcineurin inhibitors to sirolimus maintenance therapy in renal allograft recipients: 24-month efficacy and safety results from the CONVERT trial. Transplantation 2009, 87, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Mota, A.; Arias, M.; Taskinen, E.I.; Paavonen, T.; Brault, Y.; Legendre, C.; Claesson, K.; Castagneto, M.; Campistol, J.M.; Hutchinson, B.; et al. Sirolimus-based therapy flowing early cyclosporine withdrawal provides significantly improved renal histology and function at three years. Am. J. Transplant. 2004, 2, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Russ, G.; Segoloni, G.; Oberbauer, R.; Legendre, C.; Mota, A.; Eris, J.; Grinyó, J.M.; Friend, P.; Lawen, J.; Hartmann, A.; et al. Superior outcomes in renal transplantation after early cyclosporine withdrawal and sirolimus maintenance therapy, regardless of baseline renal function. Transplantation 2005, 80, 1204–1211. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Infante, B.; Schena, A.; Battaglia, M.; Ditonno, P.; Loverre, A.; Gesualdo, L.; Schena, F.P.; Grandaliano, G. Rapamycin for treatment of chronic allograft nephropathy in renal transplant patients. J. Am. Soc. Nephrol. 2005, 16, 3755–3762. [Google Scholar] [CrossRef] [PubMed]
- Lebranchu, Y.; Thierry, A.; Touopance, O.; Westeel, P.F.; Etienne, I.; Thervet, E.; Moulin, B.; Frouget, T.; Le Meur, Y.; Glotz, D.; et al. Efficacy on renal function of early conversion to sirolimus 3 months after renal transplantation: Concept study. Am. J. Transplant. 2009, 2, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Nashan, B.; Gaston, R.; Emery, V.; Säemann, M.D.; Mueller, N.J.; Couzi, L.; Dantal, J.; Shihab, F.; Mulgaonkar, S.; Seun Kim, Y.; et al. Review of cytomegalovirus infection findings with mammalian target of rapamycin inhibitor-based immunosuppressive therapy in de novo renal transplant recipients. Transplantation 2012, 93, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Tedesco-Silva, H.; Cibrik, D.; Johnston, T.; Lackova, E.; Mange, K.; Panis, C.; Walker, R.; Wang, Z.; Zibari, G.; Kim, Y.S. Everolimus plus reduced-exposure CsA versus mycopholic acid plus standard-exposure CsA in renal-transplant recipients. Am. J. Transplant. 2010, 2, 1401–1413. [Google Scholar] [CrossRef] [PubMed]
- Suwelack, B.; Malyar, V.; Koch, M.; Sester, M.; Sommerer, C. The influence of immunosuppressive agents on BK virus risk following kidney transplantation, and implications for choice of regimen. Transplant. Rev. 2012, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Legendre, C.; Campistol, J.M.; Squifflet, J.P.; Burke, J.T.; Sirolimus European Renal Transplant Study Group. Cardiovascular risk factors of sirolimus compared with cyclosporine: Early experience from two randomized trials in renal transplantation. Transplant. Proc. 2003, 35 (Suppl. 3), 151S–153S. [Google Scholar] [CrossRef]
- Joannidès, R.; Monteil, C.; de Ligny, B.H.; Westeel, P.F.; Iacob, M.; Thervet, E.; Barbier, S.; Bellien, J.; Lebranchu, Y.; Seguin, S.G.; et al. Immunosuppressant regimen based on sirolimus decreases aortic stiffness in renal transplant recipients in comparison to cyclosporine. Am. J. Transplant. 2011, 11, 2414–2422. [Google Scholar] [CrossRef] [PubMed]
- Morales, J.M. Influence of the new immunosuppressive combinations on arterial hypertension after renal transplantation. Kidney Int. Suppl. 2002, 62, S81–S87. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, E.; Marsano, L.; Bellino, D.; Cassottana, P.; Cannella, G. Effect of everolimus on left ventricular hypertrophy of de novo kidney transplant recipients: A 1 year, randomized, controlled trial. Transplantation 2012, 93, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Trinh, X.B.; Tjalma, W.A.; Vermeulen, P.B.; van den Eynden, G.; van der Auwera, I.; van Laere, S.J.; Helleman, J.; Berns, E.M.; Dirix, L.Y.; van Dam, P.A. The VEGF pathway and the AKT/mTOR/p70S6K1 signalling pathway in human epithelial ovarian cancer. Br. J. Cancer 2009, 100, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Garcia-Echeverria, C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg. Med. Chem. Lett. 2010, 20, 4308–4312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, X.F. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle 2012, 11, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Hennessy, B.T.; Slingerland, J.M. Nextgeneration mTOR inhibitors in clinical oncology: How pathway complexity informs therapeutic strategy. J. Clin. Investig. 2011, 121, 1231–1241. [Google Scholar] [CrossRef] [PubMed]
- ANZDATA Registry. 36th Report, Chapter 8: Transplantation. Australia and New Zealand Dialysis and Transplant Registry: Adelaide, Australia, 2014. Available online: http://www.anzdata.org.au (accessed on 8 March 2016).
- Ventura-Aguiar, P.; Campistol, J.M.; Diekmann, F. Safety of mTOR inhibitors in adult solid organ transplantation. Expert Opin. Drug Saf. 2016, 28, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Oberbauer, R.; Kreis, H.; Johnson, R.W.; Mota, A.; Claesson, K.; Ruiz, J.C.; Wilczek, H.; Jamieson, N.; Henriques, A.C.; Paczek, L.; et al. Long-term improvement in renal function with sirolimus after early cyclosporine withdrawal in renal transplant recipients: 2-year results of the Rapamune Maintenance Regimen Study. Transplantation 2003, 76, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Campistol, J.M.; Eris, J.; Oberbauer, R.; Friend, P.; Hutchinson, B.; Morales, J.M.; Claesson, K.; Stallone, G.; Russ, G.; Rostaing, L.; et al. Sirolimus therapy after early cyclosporine withdrawal reduces the risk for cancer in adult renal transplantation. J. Am. Soc. Nephrol. 2006, 2, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Alberú, J.; Pascoe, M.D.; Campistol, J.M.; Schena, F.P.; Rial Mdel, C.; Polinsky, M.; Neylan, J.F.; Korth-Bradley, J.; Goldberg-Alberts, R.; Maller, E.S.; et al. Lower malignancy rates in renal allograft recipients converted to sirolimus-based, calcineurin inhibitor-free immunotherapy: 24-month results from the CONVERT trial. Transplantation 2011, 92, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Maruki, Y.; Long, X.; Yoshino, K.; Oshiro, N.; Hidayat, S.; Tokunaga, C.; Avruch, J.; Yonezawa, K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002, 110, 177–189. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GβL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Thedieck, K.; Polak, P.; Kim, M.L.; Molle, K.D.; Cohen, A.; Jenö, P.; Arrieumerlou, C.; Hall, M.N. PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2007, 2, e1217. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. An expanding role for mTOR in cancer. Trends Mol. Med. 2005, 11, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Rüegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Fingar, D.C. Growing knowledge of the mTOR signaling network. Semin. Cell Dev. Biol. 2014, 36, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Facchinetti, V.; Ouyang, W.; Wei, H.; Soto, N.; Lazorchak, A.; Gould, C.; Lowry, C.; Newton, A.C.; Mao, Y.; Miao, R.Q.; et al. The mammalian target of rapamycin complex 2 controls folding and stability of Akt and protein kinase, C. EMBO J. 2008, 27, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Das, S.; Losert, W.; Parent, C.A. mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev. Cell 2010, 19, 845–857. [Google Scholar] [CrossRef] [PubMed]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-β-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125 Pt 5, 1259–1273. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Carraro, A.; Zaza, G.; Bellin, G.; Montin, U.; Violi, P.; Lupo, A.; Tedeschi, U. Epithelial to mesenchymal transition in the liver field: The double face of Everolimus in vitro. BMC Gastroenterol. 2015, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Masola, V.; Granata, S.; Bellin, G.; Dalla Gassa, A.; Onisto, M.; Gambaro, G.; Lupo, A. Sulodexide alone or in combination with low doses of everolimus inhibits the hypoxia-mediated epithelial to mesenchymal transition in human renal proximal tubular cells. J. Nephrol. 2015, 28, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Granata, S.; Gambaro, G.; Onisto, M.; Lupo, A. Everolimus-induced epithelial to mesenchymal transition in immortalized human renal proximal tubular epithelial cells: Key role of heparanase. J. Transl. Med. 2013, 11, 292. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, N.; Yoshino, K.; Hidayat, S.; Tokunaga, C.; Hara, K.; Eguchi, S.; Avruch, J.; Yonezawa, K. Dissociation of raptor from mTOR is a mechanism of rapamycin-induced inhibition of mTOR function. Genes Cells 2004, 9, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Kalinsky, K.; Heguy, A.; Bhanot, U.K.; Patil, S.; Moynahan, M.E. PIK3CA mutations rarely demonstrate genotypic intratumoral heterogeneity and are selected for in breast cancer progression. Breast Cancer Res. Treat. 2011, 129, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Pasqualetti, F.; Bocci, G.; Mey, V.; Menghini, V.; Montrone, S.; Cocuzza, P.; Ferrazza, P.; Seccia, V.; Delishaj, D.; Orlandini, C.; et al. Akt1 rs2498801 is related to survival in head and neck squamous cell cancer treated with radiotherapy. Anticancer Res. 2015, 35, 269–271. [Google Scholar] [PubMed]
- Rossi, S.; Motta, C.; Studer, V.; Monteleone, F.; de Chiara, V.; Buttari, F.; Barbieri, F.; Bernardi, G.; Battistini, L.; Cutter, G.; et al. A genetic variant of the anti-apoptotic protein Akt predicts natalizumab-induced lymphocytosis and post-natalizumab multiple sclerosis reactivation. Mult. Scler. J. 2013, 19, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Zhao, H.D.; Wang, J.L.; Huang, T.; Tian, H.W.; Yao, L.F.; Tao, H.; Chen, Z.W.; Wang, C.Y.; Sheng, S.T.; et al. Association between polymorphisms of the AKT1 gene promoter and risk of the Alzheimer’s disease in a Chinese Han Population with type 2 diabetes. CNS Neurosci. Ther. 2015, 21, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lin, L.; Xu, H.; Li, T.; Zhou, Y.; Dan, H.; Jiang, L.; Liao, G.; Zhou, M.; Li, L.; et al. Genetic variants in AKT1 gene were associated with risk and survival of OSCC in Chinese Han Population. J. Oral Pathol. Med. 2015, 44, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lin, Y.; Lan, F.; Yu, Y.; Ouyang, X.; Wang, X.; Huang, Q.; Wang, L.; Tan, J.; Zheng, F. A GG allele of 3′-side AKT1 SNP is associated with decreased AKT1 activation and better prognosis of gastric cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yamaguchi, S.; Burstein, M.D.; Terashima, K.; Chang, K.; Ng, H.K.; Nakamura, H.; He, Z.; Doddapaneni, H.; Lewis, L.; et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature 2014, 511, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yang, J.; Yu, Q.; Wu, H.; Liu, B.; Xiong, H.; Hu, G.; Zhao, J.; Yuan, X.; Liao, Z. Associations between single-nucleotide polymorphisms in the PI3K-PTEN-AKT-mTOR pathway and increased risk of brain metastasis in patients with non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 6252–6260. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Sholl, L.M.; Dal Cin, P.; Jia, Y.; Yuan, L.; MacConaill, L.; Lindeman, N.; Kuo, F.; Garcia, E.; Nucci, M.R.; et al. Targeted genomic analysis of Müllerian adenosarcoma. J. Pathol. 2015, 235, 37–49. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Witkowski, S.; Ludlow, A.T.; Roth, S.M.; Hagberg, J.M. AKT1 G205T genotype influences obesity-related metabolic phenotypes and their responses to aerobic exercise training in older Caucasians. Exp. Physiol. 2011, 96, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Pawlikowska, L.; Hu, D.; Huntsman, S.; Sung, A.; Chu, C.; Chen, J.; Joyner, A.H.; Schork, N.J.; Hsueh, W.C.; Reiner, A.P.; et al. Association of common genetic variation in the insulin/IGF1 signaling pathway with human longevity. Aging Cell 2009, 8, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Yang, H.; Hung, M.C.; Izzo, J.G.; Huang, M.; Lin, J.; Ajani, J.A.; Wu, X. Genetic variations in the PI3K/PTEN/AKT/mTOR pathway are associated with clinical outcomes in esophageal cancer patients treated with chemoradiotherapy. J. Clin. Oncol. 2009, 27, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Jones, M.R.; Chen, Y.D.; Azziz, R. First evidence of genetic association between AKT2 and polycystic ovary syndrome. Diabetes Care 2008, 31, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.E.; Ma, H.; Hale, K.S.; Yin, M.; Meyer, L.A.; Liu, H.; Li, J.; Lu, K.H.; Hennessy, B.T.; Li, X.; et al. Roles of genetic variants in the PI3K and RAS/RAF pathways in susceptibility to endometrial cancer and clinical outcomes. J. Cancer Res. Clin. Oncol. 2012, 138, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.L.; Gil, G.; Robins, H.; Hu, W.; Hirshfield, K.; Bond, E.; Bond, G.; Levine, A.J. Detection of functional single-nucleotide polymorphisms that affect apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 16297–16302. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Friedman, E.; Metsuyanim, S.; Olender, T.; Ben-Asher, E.; Papa, M.Z. Single-nucleotide polymorphisms in the p53 pathway genes modify cancer risk in BRCA1 and BRCA2 carriers of Jewish-Ashkenazi descent. Mol. Carcinog. 2010, 49, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Herrick, J.S.; Caan, B.J.; Potter, J.D.; Wolff, R.K. Diet and colorectal cancer: Analysis of a candidate pathway using SNPS, haplotypes, and multi-gene assessment. Nutr. Cancer 2011, 63, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gu, J.; Delclos, G.L.; Killary, A.M.; Fan, Z.; Hildebrandt, M.A.; Chamberlain, R.M.; Grossman, H.B.; Dinney, C.P.; Wu, X. Genetic variations of the PI3K-AKT-mTOR pathway and clinical outcome in muscle invasive and metastatic bladder cancer patients. Carcinogenesis 2010, 31, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, H.D., III; Menashe, I.; Shen, M.; Yeager, M.; Yuenger, J.; Rajaraman, P.; He, X.; Chatterjee, N.; Caporaso, N.E.; Zhu, Y.; et al. Pathway-based evaluation of 380 candidate genes and lung cancer susceptibility suggests the importance of the cell cycle pathway. Carcinogenesis 2008, 29, 1938–1943. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, J.; Greisinger, A.J.; Grossman, H.B.; Forman, M.R.; Dinney, C.P.; Hawk, E.T.; Wu, X. Energy balance, the PI3K-AKT-mTOR pathway genes, and the risk of bladder cancer. Cancer Prev. Res. 2010, 3, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Lavender, N.A.; Rogers, E.N.; Yeyeodu, S.; Rudd, J.; Hu, T.; Zhang, J.; Brock, G.N.; Kimbro, K.S.; Moore, J.H.; Hein, D.W.; et al. Interaction among apoptosis-associated sequence variants and joint effects on aggressive prostate cancer. BMC Med. Genomics 2012, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Lin, J.; Wood, C.G.; Tannir, N.M.; Wu, X. Energy balance, polymorphisms in the mTOR pathway, and renal cell carcinoma risk. J. Natl. Cancer Inst. 2013, 105, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Ellsworth, K.A.; Moon, I.; Eckloff, B.W.; Fridley, B.L.; Jenkins, G.D.; Batzler, A.; Biernacka, J.M.; Abo, R.; Brisbin, A.; Ji, Y.; et al. FKBP5 genetic variation: Association with selective serotonin reuptake inhibitor treatment outcomes in major depressive disorder. Pharmacogenet. Genom. 2013, 23, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Woillard, J.B.; Kamar, N.; Rousseau, A.; Rostaing, L.; Marquet, P.; Picard, N. Association of sirolimus adverse effects with m-TOR, p70S6K or Raptor polymorphisms in kidney transplant recipients. Pharmacogenet. Genom. 2012, 22, 725–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Tao, G.; Kang, M.; Gao, Y.; Zhu, H.; Gong, W.; Wang, M.; Wu, D.; Zhang, Z.; Zhao, Q. A polymorphism (rs2295080) in mTOR promoter region and its association with gastric cancer in a Chinese population. PLoS ONE 2013, 8, e60080. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Ju, X.; Li, P.; Meng, X.; Shao, P.; Cai, H.; Wang, M.; Zhang, Z.; Qin, C.; Yin, C. A functional variant in the MTOR promoter modulates its expression and is associated with renal cell cancer risk. PLoS ONE 2012, 7, e50302. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Wang, M.Y.; Qiu, L.X.; Zhu, M.L.; Shi, T.Y.; Zhou, X.Y.; Sun, M.H.; Yang, Y.J.; Wang, J.C.; Jin, L.; et al. Genetic variations of mTORC1 genes and risk of gastric cancer in an Eastern Chinese population. Mol. Carcinog. 2013, 52 (Suppl. 1), E70–E79. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Herrick, J.S.; Lundgreen, A.; Fitzpatrick, F.A.; Curtin, K.; Wolff, R.K. Genetic variation in a metabolic signaling pathway and colon and rectal cancer risk: mTOR, PTEN, STK11, RPKAA1, PRKAG2, TSC1, TSC2, PI3K and Akt1. Carcinogenesis 2010, 31, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, L.; Stec, R.; Cierniak, S.; Synowiec, A.; Wcisło, G.; Jesiotr, M.; Koktysz, R.; Kozłowski, W.; Szczylik, C. Clinical usefulness of PI3K/Akt/mTOR genotyping in companion with other clinical variables in metastatic renal cell carcinoma patients treated with everolimus in the second and subsequent lines. Ann. Oncol. 2015, 26, 1385–1389. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Tenorio, G.; Alkhori, L.; Olsson, B.; Waltersson, M.A.; Nordenskjöld, B.; Rutqvist, L.E.; Skoog, L.; Stål, O. PIK3CA mutations and PTEN loss correlate with similar prognostic factors and are not mutually exclusive in breast cancer. Clin. Cancer Res. 2007, 13, 3577–3584. [Google Scholar] [CrossRef] [PubMed]
- Ollikainen, M.; Gylling, A.; Puputti, M.; Nupponen, N.N.; Abdel-Rahman, W.M.; Butzow, R.; Peltomäki, P. Patterns of PIK3CA alterations in familial colorectal and endometrial carcinoma. Int. J. Cancer 2007, 121, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Wik, E.; Birkeland, E.; Trovik, J.; Werner, H.M.; Hoivik, E.A.; Mjos, S.; Krakstad, C.; Kusonmano, K.; Mauland, K.; Stefansson, I.M.; et al. High phospho-Stathmin(Serine38) expression identifies aggressive endometrial cancer and suggests an association with PI3K inhibition. Clin. Cancer Res. 2013, 19, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Tsimberidou, A.M.; Garrido-Laguna, I.; Wang, X.; Luthra, R.; Hong, D.S.; Naing, A.; Falchook, G.S.; Moroney, J.W.; Piha-Paul, S.A.; et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol. Cancer Ther. 2011, 10, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Razis, E.; Bobos, M.; Kotoula, V.; Eleftheraki, A.G.; Kalofonos, H.P.; Pavlakis, K.; Papakostas, P.; Aravantinos, G.; Rigakos, G.; Efstratiou, I.; et al. Evaluation of the association of PIK3CA mutations and PTEN loss with efficacy of trastuzumab therapy in metastatic breast cancer. Breast Cancer Res. Treat. 2011, 128, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.C.; Zhao, Y.; Zhai, R.; Liu, C.Y.; Chen, F.; Ter-Minassian, M.; Asomaning, K.; Su, L.; Heist, R.S.; Kulke, M.H.; et al. Interactions between genetic polymorphisms in the apoptotic pathway and environmental factors on esophageal adenocarcinoma risk. Carcinogenesis 2011, 32, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Agell, L.; Hernández, S.; Salido, M.; de Muga, S.; Juanpere, N.; Arumí-Uria, M.; Menendez, S.; Lorenzo, M.; Lorente, J.A.; Serrano, S.; et al. PI3K signaling pathway is activated by PIK3CA mRNA overexpression and copy gain in prostate tumors, but PIK3CA, BRAF, KRAS and AKT1 mutations are infrequent events. Mod. Pathol. 2011, 24, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Rong, M.; Grieu, F.; Iacopetta, B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Res. Treat. 2006, 96, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Mambo, E.; Guo, Z.; Hu, S.; Huang, X.; Gollin, S.M.; Trink, B.; Ladenson, P.W.; Sidransky, D.; Xing, M. Uncommon mutation, but common amplifications, of the PIK3CA gene in thyroid tumors. J. Clin. Endocrinol. Metab. 2005, 90, 4688–4693. [Google Scholar] [CrossRef] [PubMed]
- Spoerke, J.M.; O’Brien, C.; Huw, L.; Koeppen, H.; Fridlyand, J.; Brachmann, R.K.; Haverty, P.M.; Pandita, A.; Mohan, S.; Sampath, D.; et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin. Cancer Res. 2012, 18, 6771–5783. [Google Scholar] [CrossRef] [PubMed]
- Pande, M.; Bondy, M.L.; Do, K.A.; Sahin, A.A.; Ying, J.; Mills, G.B.; Thompson, P.A.; Brewster, A.M. Association between germline single nucleotide polymorphisms in the PI3K–AKT–mTOR pathway, obesity, and breast cancer disease-free survival. Breast Cancer Res. Treat. 2014, 147, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Platt, F.M.; Hurst, C.D.; Taylor, C.F.; Gregory, W.M.; Harnden, P.; Knowles, M.A. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res. 2009, 15, 6008–6017. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Stjernström, A.; Karlsson, C.; Fernandez, O.J.; Söderkvist, P.; Karlsson, M.G.; Thunell, L.K. Alterations of INPP4B, PIK3CA and pAkt of the PI3K pathway are associated with squamous cell carcinoma of the lung. Cancer Med. 2014, 3, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, M.; Nutt, C.L.; Mohapatra, G.; Louis, D.N. Genetic alterations of phosphoinositide 3-kinase subunit genes in human glioblastomas. Brain Pathol. 2004, 14, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Cui, W.; Cai, Y.; Wang, W.; Liu, Z.; Wei, P.; Bi, R.; Chen, W.; Sun, M.; Zhou, X. Frequent copy number variations of PI3K/AKT pathway and aberrant protein expressions of PI3K subunits are associated with inferior survival in diffuse large B cell lymphoma. J. Transl. Med. 2014, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, M.A.; Lippman, S.M.; Etzel, C.J.; Kim, E.; Lee, J.J.; Khuri, F.R.; Spitz, M.R.; Lotan, R.; Hong, W.K.; Wu, X. Genetic variants in the PI3K/PTEN/AKT/mTOR pathway predict head and neck cancer patient second primary tumor/recurrence risk and response to retinoid chemoprevention. Clin. Cancer Res. 2012, 18, 3705–3713. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, W.; Digon-Söntgerath, B.; Koch, A.; Waha, A.; Endl, E.; Dani, I.; Denkhaus, D.; Goodyer, C.G.; Sörensen, N.; Wiestler, O.D.; et al. Phosphatidylinositol 3′-kinase/AKT signaling is activated in medulloblastoma cell proliferation and is associated with reduced expression of PTEN. Clin. Cancer Res. 2006, 12, 3019–3027. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Furney, S.J.; Stamp, G.; Rana, S.; Ricken, G.; Oduko, Y.; Saturno, G.; Springer, C.; Hayes, A.; Gore, M.; et al. Whole-genome sequencing reveals complex mechanisms of intrinsic resistance to BRAF inhibition. Ann. Oncol. 2014, 25, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.P.; Lai, L.C.; Tsai, M.H.; Chen, P.C.; Hsu, C.P.; Lee, J.M.; Hsiao, C.K.; Chuang, E.Y. Integrated analyses of copy number variations and gene expression in lung adenocarcinoma. PLoS ONE 2011, 6, e24829. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.; Ejdelman, J.; Brimo, F.; Aprikian, A.; Chevalier, S.; Lapointe, J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer 2012, 12, 543. [Google Scholar] [CrossRef] [PubMed]
- Sircar, K.; Yoshimoto, M.; Monzon, F.A.; Koumakpayi, I.H.; Katz, R.L.; Khanna, A.; Alvarez, K.; Chen, G.; Darnel, A.D.; Aprikian, A.G.; et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009, 218, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Park, J.I.; Chae, K.S.; Kim, H.J.; Chi, S.G. Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int. J. Cancer 2003, 104, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Greco, D.; Pelttari, L.M.; Tommiska, J.; Vahteristo, P.; Heikkilä, P.; Blomqvist, C.; Aittomäki, K.; Nevanlinna, H. Variants on the promoter region of PTEN affect breast cancer progression and patient survival. Breast Cancer Res. 2011, 13, R130. [Google Scholar] [CrossRef] [PubMed]
- Hosgood, H.D., III; Menashe, I.; He, X.; Chanock, S.; Lan, Q. PTEN identified as important risk factor of chronic obstructive pulmonary disease. Respir. Med. 2009, 103, 1866–1870. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.P.; Waite, K.A.; Pilarski, R.; Hampel, H.; Fernandez, M.J.; Bos, C.; Dasouki, M.; Feldman, G.L.; Greenberg, L.A.; Ivanovich, J.; et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2003, 73, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Kurose, K.; Zhou, X.P.; Araki, T.; Cannistra, S.A.; Maher, E.R.; Eng, C. Frequent loss of PTEN expression is linked to elevated phosphorylated Akt levels, but not associated with p27 and cyclin D1 expression, in primary epithelial ovarian carcinomas. Am. J. Pathol. 2001, 158, 2097–2106. [Google Scholar] [CrossRef]
- Chen, M.; Cassidy, A.; Gu, J.; Delclos, G.L.; Zhen, F.; Yang, H.; Hildebrandt, M.A.; Lin, J.; Ye, Y.; Chamberlain, R.M.; et al. Genetic variations in PI3K-AKT-mTOR pathway and bladder cancer risk. Carcinogenesis 2009, 30, 2047–2052. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Salido, M.; Corominas, J.M.; Rojo, F.; Ferreira, B.I.; Suela, J.; Tusquets, I.; Corzo, C.; Segura, M.; Espinet, B.; et al. Are ER + PR+ and ER + PR- breast tumors genetically different? A CGH array study. Cancer Genet. 2012, 205, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.S.; Vazquez, A.; Kulkarni, D.A.; Kerrigan, J.E.; Atwal, G.; Metsugi, S.; Toppmeyer, D.L.; Levine, A.J.; Hirshfield, K.M. Polymorphic variants in TSC1 and TSC2 and their association with breast cancer phenotypes. Breast Cancer Res. Treat. 2011, 125, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, F.; Hentze, M.W. Molecular mechanisms of translational control. Nat. Rev. Mol. Cell Biol. 2004, 5, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Gingras, A.C.; Raught, B.; Sonenberg, N. eIF4 initiation factors: Effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 1999, 68, 913–963. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.A.; Blenis, J. Coordinate regulation of translation by the PI 3-kinase and mTOR pathways. Adv. Cancer Res. 2002, 86, 1–39. [Google Scholar] [PubMed]
- Avruch, J.; Belham, C.; Weng, Q.; Hara, K.; Yonezawa, K. The p70 S6 kinase integrates nutrient and growth signals to control translational capacity. Prog. Mol. Subcell. Biol. 2001, 26, 115–154. [Google Scholar] [PubMed]
- Raught, B.; Peiretti, F.; Gingras, A.C.; Livingstone, M.; Shahbazian, D.; Mayeur, G.L.; Polakiewicz, R.D.; Sonenberg, N.; Hershey, J.W. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 2004, 23, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hornstein, E.; Stolovich, M.; Levy, G.; Livingstone, M.; Templeton, D.; Avruch, J.; Meyuhas, O. Amino acid-induced translation of TOP mRNAs is fully dependent on phosphatidylinositol 3-kinase-mediated signaling, is partially inhibited by rapamycin, and is independent of S6K1 and rpS6 phosphorylation. Mol. Cell. Biol. 2001, 21, 8671–8683. [Google Scholar] [CrossRef] [PubMed]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, W.; Williams, M.; Terada, N.; Alessi, D.R.; Proud, C.G. Regulation of elongation factor 2 kinase by p90RSK1 and p70 S6 kinase. EMBO J. 2001, 20, 4370–4379. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell. Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6k1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.J.; Bröenstrup, M.; Fingar, D.C.; Jülich, K.; Ballif, B.A.; Gygi, S.; Blenis, J. SKAR is a specific target of S6 kinase 1 in cell growth control. Curr. Biol. 2004, 14, 1540–1549. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Yoon, S.O.; Richardson, C.J.; Jülich, K.; Blenis, J. SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs. Cell 2008, 133, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Beugnet, A.; Murakami, M.; Yamanaka, S.; Proud, C.G. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol. Cell. Biol. 2005, 25, 2558–2572. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Iadevaia, V.; Proud, C.G. Differing effects of rapamycin and mTOR kinase inhibitors on protein synthesis. Biochem. Soc. Trans. 2011, 39, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.; Pastore, Y.D.; Divoky, V.; Liu, E.; Mlodnicka, A.E.; Rainey, K.; Ponka, P.; Semenza, G.L.; Schumacher, A.; Prchal, J.T. Hypoxia-inducible factor-1 deficiency results in dysregulated erythropoiesis signaling and iron homeostasis in mouse development. J. Biol. Chem. 2006, 281, 25703–25711. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependant degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Using two hands to flip the angiogenic switch. Cancer Met. Rev. 2000, 19, 59–65. [Google Scholar] [CrossRef]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Cash, T.P.; Jones, R.G.; Keith, B.; Thompson, C.B.; Simon, M.C. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 2006, 21, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, J.A.; Jiang, B.H.; Iyer, N.V.; Agani, F.; Leung, S.W.; Koos, R.D.; Semenza, G.L. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 1996, 16, 4604–4013. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Ferreira, V.; Breier, G.; Pollefeyt, S.; Kieckens, L.; Gertsenstein, M.; Fahrig, M.; Vandenhoeck, A.; Harpal, K.; Eberhardt, C.; et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 1996, 380, 435–439. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am. J. Physiol. Cell Physiol. 2001, 280, C1358–C1366. [Google Scholar] [PubMed]
- Meadows, K.L.; Hurwitz, H.I. Anti-VEGF therapies in the clinic. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G.; Chen, H.; O’Connor, S.J.; Chisholm, V.; Meng, Y.G.; Krummen, L.; Winkler, M.; Ferrara, N. Humanization of an anti-VEGF monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997, 57, 4593–4599. [Google Scholar] [PubMed]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Monthouël-Kartmann, M.N.; van Obberghen, E. Regulation of hypoxia-inducible factor (HIF)-1 activity and expression of HIF hydroxylases in response to insulin-like growth factor, I. Mol. Endocrinol. 2005, 19, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Dodd, K.M.; Yang, J.; Shen, M.H.; Sampson, J.R.; Tee, A.R. mTORC1 drives HIF-1α and VEGF-A signalling via multiple mechanisms involving 4E-BP1, S6K1 and STAT3. Oncogene 2015, 34, 2239–2250. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia-inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR signaling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef]
- Pore, N.; Jiang, Z.; Gupta, A.; Cerniglia, G.; Kao, G.D.; Maity, A. EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1–independent and HIF-1-dependent mechanisms. Cancer Res. 2006, 66, 3197–3204. [Google Scholar] [CrossRef] [PubMed]
- Le Bacquer, O.; Petroulakis, E.; Paglialunga, S.; Poulin, F.; Richard, D.; Cianflone, K.; Sonenberg, N. Elevated sensitivity to diet-induced obesity and insulin resistance in mice lacking 4E-BP1 and 4E-BP2. J. Clin. Investig. 2007, 117, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.B.; Wright, H.M.; Wright, M.; Spiegelman, B.M. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 1998, 95, 4333–4337. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Chen, J. Regulation of peroxisome proliferator-activated receptor-gamma activity by mammalian target of rapamycin and amino acids in adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.T.; Ducker, G.S.; Barczak, A.J.; Barbeau, R.; Erle, D.J.; Shokat, K.M. The mammalian target of rapamycin regulates cholesterol biosynthetic gene expression and exhibits a rapamycin-resistant transcriptional profile. Proc. Natl. Acad. Sci. USA 2011, 108, 15201–15206. [Google Scholar] [CrossRef] [PubMed]
- Owen, J.L.; Zhang, Y.; Bae, S.H.; Farooqi, M.S.; Liang, G.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. USA 2012, 109, 16184–16189. [Google Scholar] [CrossRef] [PubMed]
- Kammoun, H.L.; Chabanon, H.; Hainault, I.; Luquet, S.; Magnan, C.; Koike, T.; Ferré, P.; Foufelle, F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Investig. 2009, 119, 1201–1215. [Google Scholar] [CrossRef] [PubMed]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.K.; Lee, M.Y.; Kim, J.W.; Kim, M.; Moon, J.S.; Lee, Y.J.; Ahn, Y.H.; Kim, K.S. Lipin1 is a key factor for the maturation and maintenance of adipocytes in the regulatory network with CCAAT/enhancer-binding protein α and peroxisome proliferator-activated receptor gamma 2. J. Biol. Chem. 2008, 283, 34896–34906. [Google Scholar] [CrossRef] [PubMed]
- Huffman, T.A.; Mothe-Satney, I.; Lawrence, J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. USA 2002, 99, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yabe, D.; Komuro, R.; Liang, G.; Goldstein, J.L.; Brown, M.S. Liver-specific mRNA for Insig-2 downregulated by insulin: Implications for fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2003, 100, 3155–3160. [Google Scholar] [CrossRef] [PubMed]
- Yellaturu, C.R.; Deng, X.; Park, E.A.; Raghow, R.; Elam, M.B. Insulin enhances the biogenesis of nuclear sterol regulatory element-binding protein (SREBP)-1c by posttranscriptional downregulation of Insig-2A and its dissociation from SREBP cleavage-activating protein (SCAP)·SREBP-1c complex. J. Biol. Chem. 2009, 284, 31726–31734. [Google Scholar] [CrossRef] [PubMed]
- Yellaturu, C.R.; Deng, X.; Cagen, L.M.; Wilcox, H.G.; Mansbach, C.M., II; Siddiqi, S.A.; Park, E.A.; Raghow, R.; Elam, M.B. Insulin enhances post-translational processing of nascent SREBP-1c by promoting its phosphorylation and association with COPII vesicles. J. Biol. Chem. 2009, 284, 7518–7532. [Google Scholar] [CrossRef] [PubMed]
- Bengoechea-Alonso, M.T.; Ericsson, J. A phosphorylation cascade controls the degradation of active SREBP1. J. Biol. Chem. 2009, 284, 5885–5895. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Huang, J.; Düvel, K.; Boback, B.; Wu, S.; Squillace, R.M.; Wu, C.L.; Manning, B.D. Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS ONE 2009, 4, e6189. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Park, J.; Lee, H.W.; Lee, Y.S.; Kim, J.B. Regulation of adipocyte differentiation and insulin action with rapamycin. Biochem. Biophys. Res. Commun. 2004, 321, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Ricoult, S.J.; Manning, B.D. The multifaceted role of mTORC1 in the control of lipid metabolism. EMBO Rep. 2013, 14, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Bell, A.; Grunder, L.; Sorisky, A. Rapamycin inhibits human adipocyte differentiation in primary culture. Obes. Res. 2000, 8, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, A.; Lau, S.; Sorisky, A. Rapamycin-sensitive phase of 3T3-L1 preadipocyte differentiation after clonal expansion. J. Cell. Physiol. 2001, 189, 14–22. [Google Scholar] [CrossRef] [PubMed]
- El-Chaâr, D.; Gagnon, A.; Sorisky, A. Inhibition of insulin signaling and adipogenesis by rapamycin: Effect on phosphorylation of p70 S6 kinase vs eIF4E-BP1. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Cybulski, N.; Feige, J.N.; Auwerx, J.; Ruegg, M.A.; Hall, M.N. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 2008, 8, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Yeh, W.C.; Bierer, B.E.; McKnight, S.L. Rapamycin inhibits clonal expansion and adipogenic differentiation of 3T3-L1 cells. Proc. Natl. Acad. Sci. USA 1995, 92, 11086–11090. [Google Scholar] [CrossRef] [PubMed]
- Kiberd, B.A. Cardiovascular risk reduction in renal transplantation: Strategies for success. Minerva Urol. Nefrol. 2002, 54, 51–63. [Google Scholar] [PubMed]
- Flechner, S.M.; Glyda, M.; Cockfield, S.; Grinyó, J.; Legendre, C.; Russ, G.; Steinberg, S.; Wissing, K.M.; Tai, S.S. The ORION study: Comparison of two sirolimus-based regimens versus tacrolimus and mycophenolate mofetil in renal allograft recipients. Am. J. Transplant. 2011, 11, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Brattström, C.; Wilczek, H.; Tydén, G.; Böttiger, Y.; Säwe, J.; Groth, C.G. Hyperlipidemia in renal transplant recipients treated with sirolimus (rapamycin). Transplantation 1998, 65, 1272–1274. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, H.; Bernasconi, C.; Nöldeke, J.; Yussim, A.; Mjörnstedt, L.; Erken, U.; Ketteler, M.; Navrátil, P. Cyclosporine, tacrolimus and sirolimus retain their distinct toxicity profiles despite low doses in the Symphony study. Nephrol. Dial. Transplant. 2010, 25, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Hoogeveen, R.C.; Ballantyne, C.M.; Pownall, H.J.; Opekun, A.R.; Hachey, D.L.; Jaffe, J.S.; Oppermann, S.; Kahan, B.D.; Morrisett, J.D. Effect of sirolimus on the metabolism of apoB100-containing lipoproteins in renal transplant patients. Transplantation 2001, 72, 1244–1250. [Google Scholar] [CrossRef] [PubMed]
- Morrisett, J.D.; Abdel-Fattah, G.; Hoogeveen, R.; Mitchell, E.; Ballantyne, C.M.; Pownall, H.J.; Opekun, A.R.; Jaffe, J.S.; Oppermann, S.; Kahan, B.D. Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J. Lipid Res. 2002, 43, 1170–1180. [Google Scholar] [PubMed]
- Blanchard, P.G.; Festuccia, W.T.; Houde, V.P.; St-Pierre, P.; Brûlé, S.; Turcotte, V.; Côté, M.; Bellmann, K.; Marette, A.; Deshaies, Y. Major involvement of mTOR in the PPARγ-induced stimulation of adipose tissue lipid uptake and fat accretion. J. Lipid Res. 2012, 53, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.P.; Scarpulla, R.C. Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 2004, 18, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Belandia, B.; Parker, M.G. Nuclear receptors: A rendezvous for chromatin remodeling factors. Cell 2003, 114, 277–280. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Schieke, S.M.; Phillips, D.; McCoy, J.P., Jr.; Aponte, A.M.; Shen, R.F.; Balaban, R.S.; Finkel, T. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Desai, B.N.; Myers, B.R.; Schreiber, S.L. FKBP12-rapamycin-associated protein associates with mitochondria and senses osmotic stress via mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2002, 99, 4319–4324. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA 2009, 106, 22229–22232. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [PubMed]
- Tzivion, G.; Dobson, M.; Ramakrishnan, G. FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 2011, 1813, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Cybulski, N.; Hall, M.N. TOR complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009, 34, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Wan, L.; Inuzuka, H.; Berg, A.H.; Tseng, A.; Zhai, B.; Shaik, S.; Bennett, E.; Tron, A.E.; Gasser, J.A.; et al. Rictor forms a complex with Cullin-1 to promote SGK1 ubiquitination and destruction. Mol. Cell 2010, 39, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Nada, S.; Kimura, H.; Tajima, S.; Takahashi, Y.; Kitamura, A.; Oneyama, C.; Okada, M. The mTOR pathway controls cell proliferation by regulating the FoxO3a transcription factor via SGK1 kinase. PLoS ONE 2014, 9, e88891. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [PubMed]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1·ATG13·FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed]
- Nazio, F.; Strappazzon, F.; Antonioli, M.; Bielli, P.; Cianfanelli, V.; Bordi, M.; Gretzmeier, C.; Dengjel, J.; Piacentini, M.; Fimia, G.M.; et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol. 2013, 15, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef] [PubMed]
- Filipowicz, W.; Jaskiewicz, L.; Kolb, F.A.; Pillai, R.S. Post-transcriptional gene silencing by siRNAs and miRNAs. Curr. Opin. Struct. Biol. 2005, 15, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Rukov, J.L.; Shomron, N. MicroRNA pharmacogenomics: Post-transcriptional regulation of drug response. Trends Mol. Med. 2011, 17, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Totary-Jain, H.; Sanoudou, D.; Ben-Dov, I.Z.; Dautriche, C.N.; Guarnieri, P.; Marx, S.O.; Tuschl, T.; Marks, A.R. Reprogramming of the microRNA transcriptome mediates resistance to rapamycin. J. Biol. Chem. 2013, 288, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Totary-Jain, H. Tailoring mTOR-based therapy: Molecular evidence and clinical challenges. Pharmacogenomics 2013, 14, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Chen, J.; Liu, A.; Zhou, X.; Song, Q.; Jia, C.; Chen, Z.; Lin, J.; Yang, C.; Li, M.; et al. mTORC2 promotes cell survival through c-Myc-dependent upregulation of E2F1. J. Cell Biol. 2015, 211, 105–122. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.; Zhao, Y.; Yu, L.; Xu, S.; Fu, G. MicroRNA-21 mediates the rapamycin-induced suppression of endothelial proliferation and migration. FEBS Lett. 2013, 587, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.L.; Sun, Q.S.; Liu, F.; Yang, H.W.; Liu, M.; Liu, H.X.; Xu, W.; Jiang, Y.Y. microRNA 21-mediated suppression of Sprouty1 by Pokemon affects liver cancer cell growth and proliferation. J. Cell. Biochem. 2013, 114, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, E.I.; Yousef, G.M.; Scorilas, A. Cytotoxic activity of sunitinib and everolimus in Caki-1 renal cancer cells is accompanied by modulations in the expression of apoptosis-related microRNA clusters and BCL2 family genes. Biomed. Pharmacother. 2015, 70, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ge, Y.; Drnevich, J.; Zhao, Y.; Band, M.; Chen, J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol. 2010, 189, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Xue, J.L.; Shen, Q.; Chen, J.; Tian, L. MicroRNA-7 inhibits tumor growth and metastasis by targeting the phosphoinositide 3-kinase/Akt pathway in hepatocellular carcinoma. Hepatology 2012, 55, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Zhou, H.; Zhao, H.; Zhou, Y.; Xu, R.; Xu, X.; Zheng, L.; Xue, Z.; Xia, W.; Zhang, B.; et al. MicroRNA-99a induces G1-phase cell cycle arrest and suppresses tumorigenicity in renal cell carcinoma. BMC Cancer 2012, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Fu, Q.; Lai, L.; Tao, X.; Fei, Y.; Shen, J.; Chen, Z.; Wang, Q. Downregulation of microRNA 99a in oral squamous cell carcinomas contributes to the growth and survival of oral cancer cells. Mol. Med. Rep. 2012, 6, 675–681. [Google Scholar] [PubMed]
- Oneyama, C.; Ikeda, J.; Okuzaki, D.; Suzuki, K.; Kanou, T.; Shintani, Y.; Morii, E.; Okumura, M.; Aozasa, K.; Okada, M. MicroRNA-mediated downregulation of mTOR/FGFR3 controls tumor growth induced by Src-related oncogenic pathways. Oncogene 2011, 30, 3489–3501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Liu, C.; Naji, A.; Stoffers, D.A. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic β-cells. Diabetes 2013, 62, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Iwaya, T.; Yokobori, T.; Nishida, N.; Kogo, R.; Sudo, T.; Tanaka, F.; Shibata, K.; Sawada, G.; Takahashi, Y.; Ishibashi, M.; et al. Downregulation of miR-144 is associated with colorectal cancer progression via activation of mTOR signaling pathway. Carcinogenesis 2012, 33, 2391–2397. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, A.; Kozaki, K.; Tsuruta, T.; Furuta, M.; Morita, K.; Imoto, I.; Omura, K.; Inazawa, J. The tumor suppressive microRNA miR-218 targets the mTOR component Rictor and inhibits AKT phosphorylation in oral cancer. Cancer Res. 2011, 71, 5765–5778. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, S.; Hans, F.P.; Kinniry, S.; Heinke, J.; Helbing, T.; Bluhm, F.; Sluijter, J.P.; Hoefer, I.; Pasterkamp, G.; Bode, C.; et al. MicroRNA-100 regulates neovascularization by suppression of mammalian target of rapamycin in endothelial and vascular smooth muscle cells. Circulation 2011, 123, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, A.K.; Creighton, C.J.; Yu, Z.; Zhu, H.; Gunaratne, P.H.; Reid, J.G.; Olokpa, E.; Itamochi, H.; Ueno, N.T.; Hawkins, S.M.; et al. A link between mir-100 and FRAP1/mTOR in clear cell ovarian cancer. Mol. Endocrinol. 2010, 24, 447–463. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chen, Z.; Tan, X.; Zhou, F.; Tan, F.; Gao, Y.; Sun, N.; Xu, X.; Shao, K.; He, J. MicroRNA-99a/100 promotes apoptosis by targeting mTOR in human esophageal squamous cell carcinoma. Med. Oncol. 2013, 30, 411. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Granata, S.; Tomei, P.; Dalla Gassa, A.; Lupo, A. Personalization of the immunosuppressive treatment in renal transplant recipients: The great challenge in “omics” medicine. Int. J. Mol. Sci. 2015, 16, 4281–4305. [Google Scholar] [CrossRef] [PubMed]
- Renders, L.; Frisman, M.; Ufer, M.; Mosyagin, I.; Haenisch, S.; Ott, U.; Caliebe, A.; Dechant, M.; Braun, F.; Kunzendorf, U.; et al. CYP3A5 genotype markedly influences the pharmacokinetics of tacrolimus and sirolimus in kidney transplant recipients. Clin. Pharmacol. Ther. 2007, 81, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, Y.; Djebli, N.; Szelag, J.C.; Hoizey, G.; Toupance, O.; Rérolle, J.P.; Marquet, P. CYP3A5*3 influences sirolimus oral clearance in de novo and stable renal transplant recipients. Clin. Pharmacol. Ther. 2006, 80, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Mourad, M.; Mourad, G.; Wallemacq, P.; Garrigue, V.; van Bellingen, C.; van Kerckhove, V.; de Meyer, M.; Malaise, J.; Eddour, D.C.; Lison, D.; et al. Sirolimus and tacrolimus trough concentrations and dose requirements after kidney transplantation in relation to CYP3A5 and MDR1 polymorphisms and steroids. Transplantation 2005, 80, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Granata, S.; Sallustio, F.; Grandaliano, G.; Schena, F.P. Pharmacogenomics: A new paradigm to personalize treatments in nephrology patients. Clin. Exp. Immunol. 2010, 159, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Anglicheau, D.; Le Corre, D.; Lechaton, S.; Laurent-Puig, P.; Kreis, H.; Beaune, P.; Legendre, C.; Thervet, E. Consequences of genetic polymorphisms for sirolimus requirements after renal transplant in patients on primary sirolimus therapy. Am. J. Transplant. 2005, 5, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Picard, N.; Rouguieg-Malki, K.; Kamar, N.; Rostaing, L.; Marquet, P. CYP3A5 genotype does not influence everolimus in vitro metabolism and clinical pharmacokinetics in renal transplant recipients. Transplantation 2011, 91, 652–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kniepeiss, D.; Renner, W.; Trummer, O.; Wagner, D.; Wasler, A.; Khoschsorur, G.A.; Truschnig-Wilders, M.; Tscheliessnigg, K.H. The role of CYP3A5 genotypes in dose requirements of tacrolimus and everolimus after heart transplantation. Clin. Transplant. 2011, 25, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Sam, W.J.; Chamberlain, C.E.; Lee, S.J.; Goldstein, J.A.; Hale, D.A.; Mannon, R.B.; Kirk, A.D.; Hon, Y.Y. Associations of ABCB1 3435C>T and IL-10 -1082G>A polymorphisms with long-term sirolimus dose requirements in renal transplant patients. Transplantation 2011, 92, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razzak, Z.; Loyer, P.; Fautrel, A.; Gautier, J.C.; Corcos, L.; Turlin, B.; Beaune, P.; Guillouzo, A. Cytokines downregulate expression of major cytochrome P-450 enzymes in adult human hepatocytes in primary culture. Mol. Pharmacol. 1993, 44, 707–715. [Google Scholar] [PubMed]
- Bertilsson, P.M.; Olsson, P.; Magnusson, K.E. Cytokines influence mRNA expression of cytochrome P450 3A4 and MDRI in intestinal cells. J. Pharm. Sci. 2001, 90, 638–646. [Google Scholar] [CrossRef]
- Miao, L.Y.; Huang, C.R.; Hou, J.Q.; Qian, M.-Y. Association study of ABCB1 and CYP3A5 gene polymorphisms with sirolimus trough concentration and dose requirements in Chinese renal transplant recipients. Biopharm. Drug Dispos. 2008, 29, 1–5. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Gene Symbol | Genetic Variants | Ref. | Clinical Impact |
|---|---|---|---|
| AKT1 | G49A | [56] | Association with primary breast tumor |
| rs2498804 | [57] | Association with survival and response to therapy in Squamous cell carcinoma of the head and neck | |
| rs2498804 | [58] | Association with reduced anti-apoptotic efficiency and higher risk of disease reactivation after natalizumab discontinuation in multiple sclerosis patients | |
| rs2498786 | [59] | Association with Alzheimer′s disease risk | |
| rs1130214, rs3803300; rs3730358 | [60] | Association with risk of oral squamous cell carcinoma and survival | |
| rs2498804 | [61] | Association with the risk of recurrence and survival in gastric cancer patients | |
| copy number gains of the AKT1 locus at 14q32.33 | [62] | Association with elevated mRNA expression of AKT1 in intracranial germ cell tumours | |
| rs2498804; rs2494732 | [63] | Association with risk of brain metastasis in non-small cell lung cancer | |
| A968G; G49A | [64] | SNPs found in Müllerian adenosarcoma | |
| rs1130214 | [65] | This SNP influences metabolic variables and their responses to aerobic exercise training in older, previously sedentary individuals | |
| rs3803304 | [66] | Association with lifespan | |
| rs3803304; rs2498804; rs1130214 | [67] | Association with recurrence risk, survival and response to chemoradiotherapy in esophageal cancer patients | |
| rs2498801 | [68] | Association with increased risk of endometrial cancer | |
| rs3730358; rs2498799 | [69] | Association with resistance to apoptosis contributing to the low response of caucasian EBV-transformed B lymphocyte cell lines to radiation therapy | |
| rs3730358 | [70] | Association with early age of cancer (breast and/or ovarian) onset in BRCA1/2 carriers | |
| rs3730358 | [71] | Association with lung cancer risk | |
| rs2494738 | [72] | AKT1 rs2494738 (G>A) & PDK1 rs11904366 (G>T) in combination with dietary fat and carbohydrate, influence the risk of both colon and rectal cancer | |
| AKT2 | rs892119 | [67,68] | Association with high recurrence risk and negative survival rate in esophageal cancer patients and in endometrial cancer |
| rs3730050 | [73] | Association with overall survival in metastatic bladder cancer patients | |
| rs8100018, rs3730051 | [74] | Association with polycystic ovary syndrome | |
| AKT3 | rs2994329 | [75] | Association with bladder cancer risk |
| rs2125230 | [76] | Synergistic interaction between AKT3 rs2125230-PRKCQ rs571715 and prostate cancer aggressiveness | |
| rs4132509; rs3766673; rs12031994; rs4430311; rs1058304; rs2345994 | [77] | Association with increased risk of renal cell carcinoma | |
| FKBP5 | rs352428 | [78] | Association with a decreased transcriptional activity and low FKBP5 expression resulting in poor response to serotonin reuptake inhibitors in patients with major depressive disorder |
| MTOR | G6981T; T4358C; A6139T; A5941G; T6643C | [62] | Mutations found in intracranial germ cell tumours |
| rs11121704; rs2295080 | [67] | Association with poor survival and poor response to taxane in esophageal cancer patients | |
| AGAAA haplotype (rs1770345/rs2300095/rs2076655/rs1883965/rs12732063) | [79] | This haplotype, in addition to SRL trough levels, was significantly associated with a decrease in haemoglobin levels in renal transplant recipients switched from a calcineurin inhibitor to sirolimus | |
| rs2295080 | [80,81] | Association with gastric cancer risk and renal cell carcinoma susceptibility by modulating the endogenous MTOR expression level | |
| rs1883965 | [82] | Association with an increased risk of gastric cancer | |
| rs2024627; rs1057079 | [83] | Association with colon cancer | |
| PI3KCA | A3140G; G1633A; G1624A, A3140T; T1035A; A1637C; G1633C | [56] | Association with primary breast tumor |
| rs7621329 | [61] | Association with the risk of recurrence in gastric cancer patients | |
| rs2699887 | [63] | Association with risk of brain metastasis in non-small cell lung cancer | |
| rs6443624; rs9838411; rs2699887 | [68] | Association with risk, survival and recurrence of endometrial cancer | |
| rs6443624 | [84] | Association with survival in renal cell carcinoma patients treated with everolimus | |
| G1624A; G1633A; A1637C; A3140G; A3062G; G3145C; A3140G | [85] | These mutations are very common in breast cancer and associated with estrogen receptor(+) status, small size and the risk to relapse | |
| del325-327; gene amplification; A1634G; G1633A; A1634C; A3140T; A3140G; T335A; G1638T; A3062C; C3074A; T3107C; A3140G | [86] | A1634G; G1633A; A1634C; A3140T; A3140G are mutations found in Colorectal cancer. T335A; G1638T; A3062C; C3074A; T3107C; A3140G; T3141G are mutations found in endometrial carcinomas | |
| amplification of the 3q26 region, increased PIK3CA copy number | [87] | Association with high pStathmin(S38) level, a marker of poor prognosis in endometrial cancer patients | |
| C112T; G113A; G263A; C311G; G317T; G323C; del332-334; G353A; G365A; C370A; G1048C; T1132C; T1258C; G1357C; C1616G; G1624A; A1625G; A1625T; G1633A; A1634G; G1635T; C1636A; A1637C; C1981A; A2102C; G2702T; T3022C; A3073G; C3074A; G3129T; C3139T; A3140G; A3140T; G3145A | [88] | These mutations have been found in several human cancers | |
| A3140G; G1624A; C1636A; G1633A; G3145A; G1645A; G3129C | [89] | These mutations are highly frequent in patients with endometrial, ovarian, colorectal, breast, cervical cancer, NSCLC, and squamous cell cancer of head and neck. The response rate was significantly higher for patients with PIK3CA mutations treated with PI3K/AKT/mTOR pathway inhibitors | |
| G1624A; G1633A; A3140G | [90] | Association with worse time to progression in the HER2-positive patients with metastatic breast cancer treated with Trastuzumab | |
| PI3KCA | rs4855094, rs7644468 | [91] | Subjects carrying the variant allele of rs4855094 or rs7644468 significantly enhanced the risk of gastroesophageal reflux disease to develop esophageal adenocarcinoma compared with subjects carrying homozygous wild genotypes |
| IVS9+91 | [92] | Mutation found in prostate tumours | |
| rs7651265 | [83] | Association with rectal cancer | |
| C1241T; T1258C; del1352–1366; G1624A; G1633A; A1634G; C1636A; C1636G; A3140G; A3140T | [93] | Association with breast tumors and with significantly worse survival | |
| C3075T; gene amplification | [94] | These mutations have been found in thyroid cancer | |
| gene amplification; G1624A; G1633A; G353A; A331G | [95] | Mutations found in non-small-cell lung cancer | |
| rs2677760 | [96] | This SNP was strongly associated with worse breast cancer disease-free survival in the overweight and obese patients | |
| G1624A; G1633A; A1928G; G3129A; A3140G | [97] | These SNPs have been found in bladder cancer | |
| G1633A | [98,99] | Mutation found in pancreatic neuroendocrine tumors and in squamous cell carcinoma | |
| Copy number variations | [100,101] | Amplification found in glioblastoma. Copy number variations found in diffuse large B-cell lymphoma had significantly shorter survival times | |
| PIK3CB | Copy number variations | [101] | Association with significantly shorter survival times in diffuse large B-cell lymphoma |
| PIK3C2B | N232del; A577S; A577S | [64] | SNPs found in Müllerian adenosarcoma |
| PIK3CD | rs4129341 | [102] | Association with a high risk to develop second primary tumors in patients with head and neck squamous cell carcinoma. The same variant genotype was also associated with significant benefit following 13-cis-Retinoic acid intervention |
| Copy number variations | [100] | Found in Glioblastoma | |
| PIK3C2G | T3130C | [62] | SNP found in intracranial germ cell tumours |
| PRKCQ | rs571715 | [76] | Interactions between AKT3 rs12031994-PRKCQ rs571715 as well as AKT3 rs12031994-BID rs366542-PRKCQ rs571715 were significantly associated with disease aggressiveness in prostate cancer |
| PIK3R1 | 9-bp del | [100] | Mutation found in glioblastoma |
| rs1862162 | [68] | Association with risk of endometrial cancer and the hazard of death | |
| rs10515074 | [73] | Association with survival in muscle invasive and metastatic bladder cancer patients | |
| PTEN | rs701848 | [61,81] | Association with the risk of recurrence and survival in gastric cancer patients and with an increased renal cell carcinoma risk |
| G407A | [62] | SNP found in intracranial germ cell tumours | |
| rs12357281 | [67] | Association with a decreased recurrence risk of esophageal cancer | |
| rs532678 | [72] | This SNP, in association with PDK1 rs11904366 (G>T), PRKAG2 rs1881632 (C>T) and dietary fat and carbohydrate, influence the risk of both colon and rectal cancer | |
| gene loss | [85] | PTEN loss by itself or combined with mutated PIK3CA tended to confer radiosensitivity in breast cancer patients | |
| gene loss | [86] | PTEN protein expression was more often decreased or lost in endometrial carcinomas than colorectal cancer | |
| gene loss | [90] | Association with increased risk of death in the HER2-positive patients with metastatic breast cancer treated with Trastuzumab | |
| gene loss | [95] | PTEN loss was observed in non-small-cell lung cancer tumor samples with both squamous cell and adenocarcinoma histologies and render the cells sensitive to the PI3K inhibitor GDC-0941 | |
| 738delG; T323G; 961_962insTGACAAGGAATATCTAGTACTTACTTTAA; T202C; G494A | [98] | Mutations found in pancreatic neuroendocrine tumors | |
| R130X; L139X; R142Q; delAAGCT (codon 125-126); G165E; delAGAA (codon 183-184); delCCCT (codon 319-320) | [99] | Mutations found in squamous cell carcinoma and adenocarcinoma. Some are associated with loss of PTEN | |
| 34-bp insertion in exon 7, a 4-bp deletion in exon 8, a 1-bp insertion in exon 7 and a point mutation in intron 3 | [100] | Mutations found in glioblastoma | |
| rs1234221 | [102] | Association with an high risk to develop second primary tumors in patients with head and neck squamous cell carcinoma and with significant benefit following 13-cis-Retinoic acid intervention | |
| gene loss | [103] | PTEN mRNA and protein levels were found to be significantly lower in medulloblastomas compared with normal cerebellar tissue of different developmental stages | |
| PTEN frame-shift deletion | [104] | Association with AKT hyper-activation in melanoma | |
| copy number variations | [105] | Association with lung tumorigenesis | |
| deletion | [106,107] | Association with early disease recurrence, reduced levels of androgen receptor expression and pAKT activation in prostate cancer | |
| deletion | [108] | Association with gastric carcinogenesis | |
| PTEN | promoter polymorphisms (-903GA, -975GC, and -1026CA) | [109] | Association with worse long term survival and risk of distant metastasis in breast cancer patients |
| rs701848; rs1903858 | [110] | Association with decreased chronic obstructive pulmonary disease risk | |
| Deletion homozygosity (from D10S1765 to D10S541; from D10S215 to IVS4+109; from D10S215 to IVS8+32). Promoter region (1238A/G; 1110A/G; 1084C/T; 1000T/C; 930G/A; 920G/T; 895A/C; 861G/T; 834C/T; 764G/A) | [111] | Patients carrying the promoter mutations or deletions showed a decrease in PTEN protein of the correct molecular weight with nonfunctional lipid phosphatase activity and elevated level of phosphorylated Akt in patients with Cowden syndrome and patients with Bannayan-Riley-Ruvalcaba syndrome | |
| IVS1+41C>G; c.166T>G; c.70G>T; c.463T>A; 469–470insG; 741–742insA; c.862G>T; IVS3-1G>T; allelic loss | [112] | Association with reduced or absent PTEN protein expression in primary adenocarcinomas of the ovary | |
| RAPTOR | rs9906827; rs7208502 | [73] | Association with survival in metastatic bladder cancer patients |
| rs11653499; rs7212142; rs7211818; rs7208536; rs4969444; rs2048753; rs2672890; rs9897968; rs1877926; rs2271612; rs6420481; rs1062935 | [75] | Association with bladder cancer risk | |
| rs11653499, rs7211818, rs7212142; rs9674559 | [113] | Association with bladder cancer risk | |
| RHEB | rs717775 | [75] | Association with bladder cancer risk |
| RPS6KA5 | rs7155799 | [75] | Association with bladder cancer risk |
| RPS6KB1 | gained regions | [14] | This gene was highly amplified in estrogen receptor (ER)+/progesterone receptor (PR)− breast tumors compared with ER+PR+ tumors |
| TSC1 | rs2519757 | [96] | Association with improved disease-free survival in breast cancer |
| rs7040593; rs3827665; rs739442; rs2519760; rs2809243; rs4962225; rs7035940; rs10491534; rs2073869; rs7874234; rs869116; rs4367688 | [102] | Association with a high risk to develop second primary tumors in patients with head and neck squamous cell carcinoma. Rs739442, rs4962225 and rs7874234 were also associated with significant benefit following 13-cis-Retinoic acid intervention | |
| 73-77Δ5; C104G; C163T; A203G; A314G; T473G; C555G; C585A; C616G; T648A; IVS7-1G>A; C866A; G1041A; C1250T; 1531insA; C1579T; 1727-1748Δ22insG; 1872ΔT; 1958-1959ΔTA; C2612G; IVS20+1G>A; C2851T | [97] | These mutations are common in bladder cancer | |
| rs13295634; rs11243940 | [72] | PDK1 rs11904366 (G>T) & TSC1 rs11243940 (A>G) in combination with dietary fat and carbohydrate, influence the risk of both colon and rectal cancer. | |
| rs7874234 | [83] | Association with a significant 40% reduction in the risk of rectal and colon cancer | |
| rs7874234 | [114] | Association with age at diagnosis in ductal estrogen receptor (ER)+ breast carcinoma patients | |
| TSC2 | rs2073636 | [75] | Association with bladder cancer risk |
| p.A1429S; p.F1510del | [64] | Mutations found in Müllerian adenosarcoma | |
| rs3087631 | [83] | Association with colon cancer | |
| C3422T; G4498A; 4113_4114delTG; C5383T; C26A; A4952G | [98] | These mutations are common in pancreatic neuroendocrine tumors | |
| rs13335638 | [115] | Association with breast cancer |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granata, S.; Dalla Gassa, A.; Carraro, A.; Brunelli, M.; Stallone, G.; Lupo, A.; Zaza, G. Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects. Int. J. Mol. Sci. 2016, 17, 735. https://doi.org/10.3390/ijms17050735
Granata S, Dalla Gassa A, Carraro A, Brunelli M, Stallone G, Lupo A, Zaza G. Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects. International Journal of Molecular Sciences. 2016; 17(5):735. https://doi.org/10.3390/ijms17050735
Chicago/Turabian StyleGranata, Simona, Alessandra Dalla Gassa, Amedeo Carraro, Matteo Brunelli, Giovanni Stallone, Antonio Lupo, and Gianluigi Zaza. 2016. "Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects" International Journal of Molecular Sciences 17, no. 5: 735. https://doi.org/10.3390/ijms17050735
APA StyleGranata, S., Dalla Gassa, A., Carraro, A., Brunelli, M., Stallone, G., Lupo, A., & Zaza, G. (2016). Sirolimus and Everolimus Pathway: Reviewing Candidate Genes Influencing Their Intracellular Effects. International Journal of Molecular Sciences, 17(5), 735. https://doi.org/10.3390/ijms17050735