
Dendritic-Tumor Fusion Cell-Based Cancer Vaccines


Abstract
:1. Introduction
1.1. Dendritic Cell (DC)-Based Cancer Vaccines
1.2. Fusions of Autologous DCs and Autologous Whole Tumor Cells
1.3. Fusion of Autologous DCs and Allogeneic Whole Tumor Cells
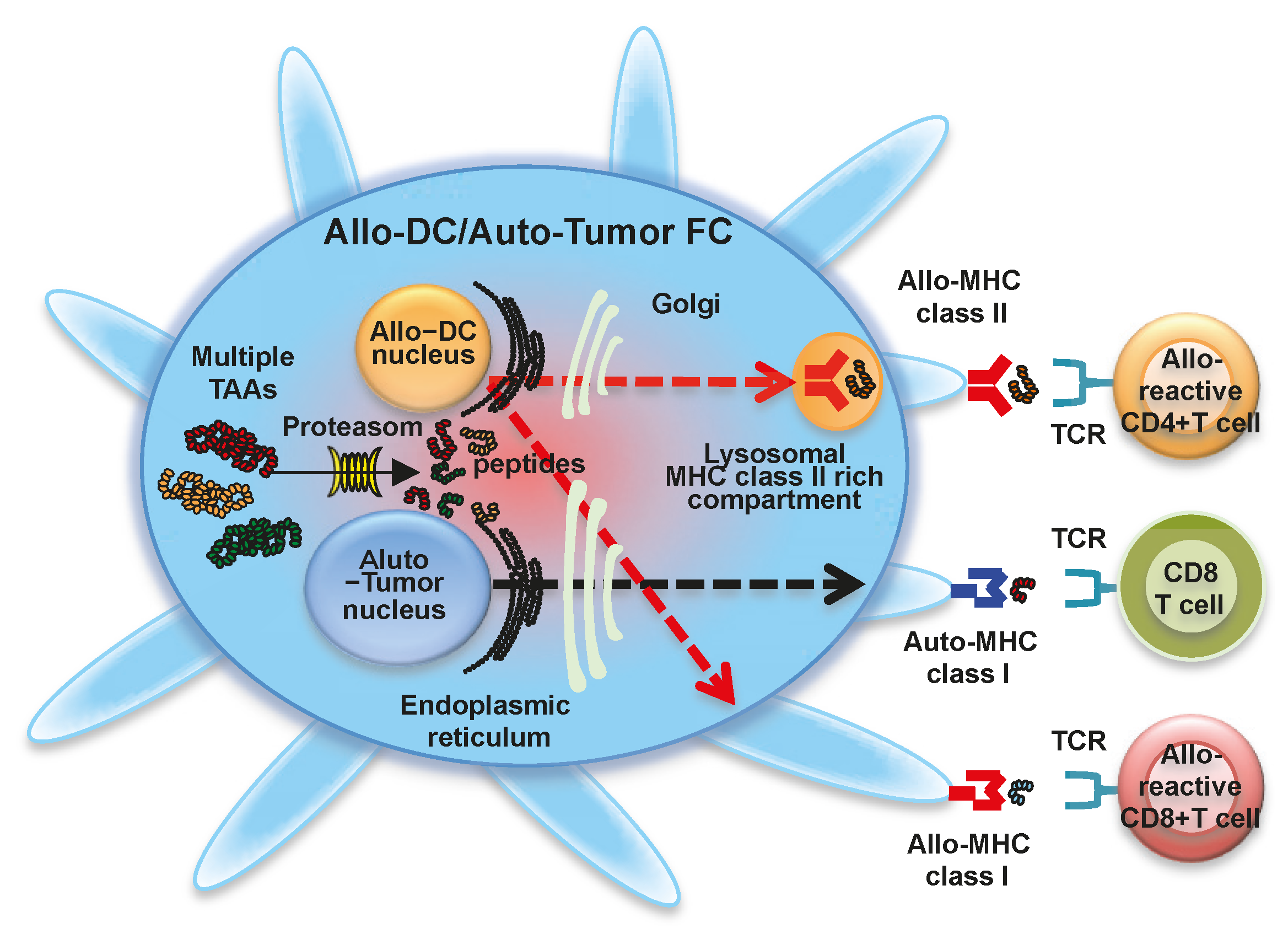
1.4. Fusion of Allogeneic DCs and Autologous Whole Tumor Cells
1.5. Fusion of Allogeneic DCs and Allogeneic Whole Tumor Cells
1.6. Fusion of Activated Autologous DCs and Autologous Whole Tumor Cells
1.7. Fusion of Autologous DCs and Immunogenic Autologous Whole Tumor Cells
1.8. Fusion of DCs and Cancer Stem Cells
1.9. Chaperone-Peptide Complexes from DC-Tumor Fusion Cells (FCs)
1.10. DC-Tumor FC Combination Therapy
1.11. Future Cancer Regimens Using DC-Tumor FCs
2. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| APCs | Antigen presenting cells |
| CRT | Calreticulin |
| CSCs | Cancer stem cells |
| CTLs | Cytotoxic T lymphocytes |
| DCs | Dendritic cells |
| DC-tumor FCs | Fusions of DCs and whole tumor cells |
| GMP | Good manufacturing practice |
| GMP | Granulocyte macrophage colony-stimulating factor |
| HSPs | Heat shock proteins |
| HSP70.PC | HSP70-peptide complexes |
| HSP70.PC-FCs | HSP70.PC from DC-tumor FCs |
| HSP70.PC-T | HSP70.PC from whole tumor cells |
| IFN-γ | Interferon-γ |
| IL | Interleukin |
| iNKT | Invariant natural killer T |
| MHC | Major histocompatibility complex |
| MDSCs | Myeloid derived suppresser cells |
| NK | Natural killer |
| Poly(I:C) | Polyriboinosinic polyribocytidylic acid |
| PEG | Polyethylene glycol |
| PD1 | Programmed death 1 |
| PD-L1 | Programmed death ligand 1 |
| PGE2 | Prostaglandin E2 |
| Tregs | Regulatory T cells |
| Th1 | T helper type 1 |
| TLR | Toll-like receptor |
| TGF-β | Transforming growth factor β |
| TAAs | Tumor-associated antigens |
| TNF-α | Tumor necrosis factor-α |
| VEGF | Vascular endothelial growth factor |
References
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Swanson, J. The endocytic activity of dendritic cells. J. Exp. Med. 1995, 182, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bloy, N.; Pol, J.; Aranda, F.; Eggermont, A.; Cremer, I.; Fridman, W.H.; Fučíková, J.; Galon, J.; Tartour, E.; Spisek, R.; et al. Trial watch: Dendritic cell-based anticancer therapy. Oncoimmunology 2014, 3, e963424. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, J.; Bourgeois-Daigneault, M.; Lapointe, R. Targeting the MHC class II antigen presentation pathway in cancer immunotherapy. Oncoimmunology 2012, 1, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.J.; Wimmers, F.; Hato, S.V.; de Vries, I.J.; Sköld, A.E. Dendritic cell cross talk with innate and innate-like effector cells in antitumor immunity: Implications for DC vaccination. Crit. Rev. Immunol. 2014, 34, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; King, S.; Allgeier, T.; Braumüller, H.; Lüking, C.; Mysliwietz, J.; Kriegeskorte, A.; Busch, D.H.; Röcken, M.; Mocikat, R. DC-NK cell cross talk as a novel CD4+ T-cell-independent pathway for antitumor CTL induction. Blood 2005, 106, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Wehner, R.; Dietze, K.; Bachmann, M.; Schmitz, M. The bidirectional crosstalk between human dendritic cells and natural killer cells. J. Innate Immun. 2011, 3, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Takakura, K.; Mori, M.; Yoshizaki, S.; Tsukinaga, S.; Odahara, S.; Koyama, S.; Imazu, H.; et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin. Cancer Res. 2014, 20, 4228–4239. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Nagaoka, K.; Takahara, M.; Yang, X.Y.; Liu, C.X.; Guo, H.; Roy Choudhury, K.; Hobeika, A.; Hartman, Z.; Morse, M.A.; et al. Precision cancer immunotherapy: Optimizing dendritic cell-based strategies to induce tumor antigen-specific T-cell responses against individual patient tumors. J. Immunother. 2015, 38, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Nestle, F.O.; Alijagic, S.; Gilliet, M.; Sun, Y.; Grabbe, S.; Dummer, R.; Burg, G.; Schadendorf, D. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 1998, 4, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Bloy, N.; Buqué, A.; Eggermont, A.; Cremer, I.; Sautès-Fridman, C.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; et al. Trial watch: Peptide-based anticancer vaccines. Oncoimmunology 2015, 4, e974411. [Google Scholar] [CrossRef] [PubMed]
- Kanodia, S.; Kast, W.M. Peptide-based vaccines for cancer: Realizing their potential. Expert Rev. Vaccines 2008, 7, 1533–1545. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Immunotherapy: Past, present and future. Nat. Med. 2003, 9, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Ueno, H.; Connolly, J.; Kerneis-Norvell, F.; Blanck, J.P.; Johnston, D.A.; Fay, J.; Banchereau, J. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and Mart-1 specific CD8+ T-cell immunity. J. Immunother. 2006, 29, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, E.; Nair, S.K.; Lyerly, H.K. Immunotherapy of cancer with dendritic-cell-based vaccines. Cancer Immunol. Immunother. 1998, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Bubeník, J. Genetically engineered dendritic cell-based cancer vaccines (Review). Int. J. Oncol. 2001, 18, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, L.; Chen, D.; Kashiwaba, M.; Manome, Y.; Tanaka, T.; Kufe, D. Induction of antigen-specific antitumor immunity with adenovirus-transduced dendritic cells. Gene Ther. 1997, 4, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Ohana, M.; Liu, C.; Nikrui, N.; Durfee, J.; Lerner, A.; Gong, J. Dendritic cells fused with human cancer cells: Morphology, antigen expression, and T cell stimulation. Clin. Immunol. 2004, 113, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Koido, S.; Calderwood, S.K. Cell fusion: From hybridoma to dendritic cell-based vaccine. Expert Rev. Vaccines 2008, 7, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, M.; Takakura, K.; Ohkusa, T.; Koido, S. The impact of dendritic cell-tumor fusion cells on cancer vaccines—Past progress and future strategies. Immunotherapy 2015, 7, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Takakura, K.; Kajihara, M.; Ito, Z.; Ohkusa, T.; Gong, J.; Koido, S. Dendritic-tumor fusion cells in cancer immunotherapy. Discov. Med. 2015, 19, 169–174. [Google Scholar] [PubMed]
- Koido, S.; Gong, J. Characterization of structure and direct antigen presentation by dendritic/tumor-fused cells as cancer vaccines. Anticancer Res. 2013, 33, 347–354. [Google Scholar] [PubMed]
- Koido, S.; Gong, J. Cell fusion between dendritic cells and whole tumor cells. Methods Mol. Biol. 2015, 1313, 185–191. [Google Scholar] [PubMed]
- Koido, S.; Tanaka, Y.; Chen, D.; Kufe, D.; Gong, J. The kinetics of in vivo priming of CD4 and CD8 T cells by dendritic/tumor fusion cells in MUC1-transgenic mice. J. Immunol. 2002, 168, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Nikrui, N.; Ohana, M.; Xia, J.; Tanaka, Y.; Liu, C.; Durfee, J.K.; Lerner, A.; Gong, J. Assessment of fusion cells from patient-derived ovarian carcinoma cells and dendritic cells as a vaccine for clinical use. Gynecol. Oncol. 2005, 99, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Torii, A.; Homma, S.; Toyama, Y.; Kawahara, H.; Ogawa, M.; Watanabe, M.; Yanaga, K.; Fujise, K.; et al. Induction of antigen-specific CD4- and CD8-mediated T-cell responses by fusions of autologous dendritic cells and metastatic colorectal cancer cells. Int. J. Cancer 2005, 117, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Fujise, K.; Gong, J.; Tajiri, H. Dendritic/tumor fusion cell-based vaccination against cancer. Arch. Immunol. Ther. Exp. 2007, 55, 281–287. [Google Scholar] [CrossRef]
- Koido, S.; Homma, S.; Hara, E.; Namiki, Y.; Takahara, A.; Komita, H.; Nagasaki, E.; Ito, M.; Ohkusa, T.; Gong, J.; et al. Regulation of tumor immunity by tumor/dendritic cell fusions. Clin. Dev. Immunol. 2010, 2010, 516768. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Fusions between dendritic cells and whole tumor cells as anticancer vaccines. Oncoimmunology 2013, 2, e24437. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Enomoto, Y.; Apostolopoulos, V.; Gong, J. Tumor regression by CD4 T-cells primed with dendritic/tumor fusion cell vaccines. Anticancer Res. 2014, 34, 3917–3924. [Google Scholar] [PubMed]
- Tanaka, Y.; Koido, S.; Ohana, M.; Liu, C.; Gong, J. Induction of impaired antitumor immunity by fusion of MHC class II-deficient dendritic cells with tumor cells. J. Immunol. 2005, 174, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Torii, A.; Toyama, Y.; Kawahara, H.; Watanabe, M.; Yanaga, K.; Fujise, K.; Tajiri, H.; et al. Dendritic cells fused with allogeneic colorectal cancer cell line present multiple colorectal cancer-specific antigens and induce antitumor immunity against autologous tumor cells. Clin. Cancer Res. 2005, 11, 7891–7900. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Tanaka, Y.; Tajiri, H.; Gong, J. Generation and functional assessment of antigen-specific T cells stimulated by fusions of dendritic cells and allogeneic breast cancer cells. Vaccine 2007, 25, 2610–2619. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Namiki, Y.; Komita, H.; Takahara, A.; Nagasaki, E.; Ito, M.; Sagawa, Y.; Mitsunaga, M.; et al. Dendritic/pancreatic carcinoma fusions for clinical use: Comparative functional analysis of healthy- versus patient-derived fusions. Clin. Immunol. 2010, 135, 384–400. [Google Scholar] [CrossRef] [PubMed]
- de Gruijl, T.D.; van den Eertwegh, A.J.; Pinedo, H.M.; Scheper, R.J. Whole-cell cancer vaccination: From autologous to allogeneic tumor- and dendritic cell-based vaccines. Cancer Immunol. Immunother. 2008, 57, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Fabre, J.W. The allogeneic response and tumor immunity. Nat. Med. 2001, 7, 649–652. [Google Scholar] [CrossRef] [PubMed]
- atthaporn, S.; Robins, A.; Vassanasiri, W.; El-Sheemy, M.; Jibril, J.A.; Clark, D.; Valerio, D.; Eremin, O. Dendritic cells are dysfunctional in patients with operable breast cancer. Cancer Immunol. Immunother. 2004, 53, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Ohkusa, T.; Gong, J.; Tajiri, H. Cancer immunotherapy by fusions of dendritic cells and tumor cells. Immunotherapy 2009, 1, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Koido, S.; Chen, D.; Gendler, S.J.; Kufe, D.; Gong, J. Vaccination with allogeneic dendritic cells fused to carcinoma cells induces antitumor immunity in MUC1 transgenic mice. Clin. Immunol. 2001, 101, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Kamigaki, T.; Kawasaki, K.; Nakamura, T.; Yamamoto, M.; Kanemitsu, K.; Takase, S.; Kuroda, D.; Kim, Y.; Ajiki, T.; et al. Superior anti-tumor protection and therapeutic efficacy of vaccination with allogeneic and semiallogeneic dendritic cell/tumor cell fusion hybrids for murine colon adenocarcinoma. Cancer Immunol. Immunother. 2007, 56, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.W.; Cowled, C.J.; Darling, D.; Guinn, B.A.; Farzaneh, F.; Noble, A.; Galea-Lauri, J. Semi-allogeneic dendritic cells can induce antigen-specific T-cell activation, which is not enhanced by concurrent alloreactivity. Cancer Immunol. Immunother. 2007, 56, 1861–1873. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Nikrui, N.; Chen, D.; Koido, S.; Wu, Z.; Tanaka, Y.; Cannistra, S.; Avigan, D.; Kufe, D. Fusions of human ovarian carcinoma cells with autologous or allogeneic dendritic cells induce antitumor immunity. J. Immunol. 2000, 165, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D.E.; Vasir, B.; George, D.J.; Oh, W.K.; Atkins, M.B.; McDermott, D.F.; Kantoff, P.W.; Figlin, R.A.; Vasconcelles, M.J.; Xu, Y.; et al. Phase I/II study of vaccination with electrofused allogeneic dendritic cells/autologous tumor-derived cells in patients with stage IV renal cell carcinoma. J. Immunother. 2007, 30, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Märten, A.; Renoth, S.; Heinicke, T.; Albers, P.; Pauli, A.; Mey, U.; Caspari, R.; Flieger, D.; Hanfland, P.; von Ruecker, A.; et al. Allogeneic dendritic cells fused with tumor cells: Preclinical results and outcome of a clinical phase I/II trial in patients with metastatic renal cell carcinoma. Hum. Gene Ther. 2003, 14, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Kan, S.; Takakura, K.; Namiki, Y.; Kobayashi, H.; Ito, Z.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; et al. Induction of antigen-specific cytotoxic T lymphocytes by fusion cells generated from allogeneic plasmacytoid dendritic and tumor cells. Int. J. Oncol. 2014, 45, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Siders, W.M.; Garron, C.; Shields, J.; Kaplan, J.M. Induction of antitumor immunity by semi-allogeneic and fully allogeneic electrofusion products of tumor cells and dendritic cells. Clin. Transl. Sci. 2009, 2, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Namiki, Y.; Ohkusa, T.; Gong, J.; Tajiri, H. Cancer vaccine by fusions of dendritic and cancer cells. Clin. Dev. Immunol. 2009, 2009, 657369. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, F.; Iinuma, H.; Okinaga, K. Dendritic cell vaccine therapy by immunization with fusion cells of interleukin-2 gene-transduced, spleen-derived dendritic cells and tumour cells. Scand. J. Immunol. 2004, 59, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Koido, S.; Chen, D.; Tanaka, Y.; Huang, L.; Avigan, D.; Anderson, K.; Ohno, T.; Kufe, D. Immunization against murine multiple myeloma with fusions of dendritic and plasmacytoma cells is potentiated by interleukin 12. Blood 2002, 12, 2512–2517. [Google Scholar] [CrossRef]
- Iinuma, T.; Homma, S.; Noda, T.; Kufe, D.; Ohno, T.; Toda, G. Prevention of gastrointestinal tumors based on adenomatous polyposis coli gene mutation by dendritic cell vaccine. J. Clin. Investig. 2004, 113, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Iinuma, H.; Okinaga, K.; Fukushima, R.; Inaba, T.; Iwasaki, K.; Okinaga, A.; Takahashi, I.; Kaneko, M. Superior protective and therapeutic effects of IL-12 and IL-18 gene-transduced dendritic neuroblastoma fusion cells on liver metastasis of murine neuroblastoma. J. Immunol. 2006, 176, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Vasir, B.; Wu, Z.; Crawford, K.; Rosenblatt, J.; Zarwan, C.; Bissonnette, A.; Kufe, D.; Avigan, D. Fusions of dendritic cells with breast carcinoma stimulate the expansion of regulatory T cells while concomitant exposure to IL-12, CpG oligodeoxynucleotides, and anti-CD3/CD28 promotes the expansion of activated tumor reactive cells. J. Immunol. 2008, 181, 808–821. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, K.; Yamamoto, S.; Otsuru, S.; Nakai, S.; Tamai, K.; Morishita, R.; Ogihara, T.; Kaneda, Y. Enhanced tumor-specific long-term immunity of hemagglutinating [correction of hemaggluttinating] virus of Japan-mediated dendritic cell-tumor fused cell vaccination by coadministration with CpG oligodeoxynucleotides. J. Immunol. 2004, 173, 4297–4307. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Cai, S.; Liu, P.; Zeng, J.; He, Y.; Wu, X.; Du, J. Enhancement of dendritic cell-tumor fusion vaccine potency by indoleamine-pyrrole 2,3-dioxygenase inhibitor, 1-MT. J. Cancer Res. Clin. Oncol. 2008, 134, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Kikuchi, T.; Irie, M.; Yamamoto, Y.; Arai, T.; Tanaka, T.; Joki, T.; Abe, T. Cotransfection of poly(I: C) and siRNA of IL-10 into fusions of dendritic and glioma cells enhances antitumor T helper type 1 induction in patients with glioma. J. Immunother. 2011, 34, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Abe, T.; Fukuda, T.; Saotome, H.; Ryan, J.L.; Kufe, D.W.; Ohno, T. Vaccination of glioma patients with fusions of dendritic and glioma cells and recombinant human interleukin 12. J. Immunother. 2004, 27, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, T.; Akasaki, Y.; Irie, M.; Homma, S.; Abe, T.; Ohno, T. Results of a phase I clinical trial of vaccination of glioma patients with fusions of dendritic and glioma cells. Cancer Immunol. Immunother. 2001, 50, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a promising candidate for tumor immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419–435. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Pearce, E.J. Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol. 2014, 5, 203. [Google Scholar] [CrossRef] [PubMed]
- Warger, T.; Osterloh, P.; Rechtsteiner, G.; Fassbender, M.; Heib, V.; Schmid, B.; Schmitt, E.; Schild, H.; Radsak, M.P. Synergistic activation of dendritic cells by combined toll-like receptor ligation induces superior CTL responses in vivo. Blood 2006, 108, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Napolitani, G.; Rinaldi, A.; Bertoni, F.; Sallusto, F.; Lanzavecchia, A. Selected toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 2005, 6, 769–776. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Hara, E.; Homma, S.; Mitsunaga, M.; Takahara, A.; Nagasaki, E.; Kawahara, H.; Watanabe, M.; Toyama, Y.; Yanagisawa, S.; et al. Synergistic induction of antigen-specific CTL by fusions of TLR-stimulated dendritic cells and heat-stressed tumor cells. J. Immunol. 2007, 179, 4874–4883. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Takahara, A.; Odahara, S.; Tsukinaga, S.; Yukawa, T.; Mitobe, J.; et al. Combined TLR2/4-activated dendritic/tumor cell fusions induce augmented cytotoxic T lymphocytes. PLoS ONE 2013, 8, e59280. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Gilmour, J.W.; Imami, N.; Amjadi, P.; Henderson, D.C.; Allen-Mersh, T.G. Increased serum transforming growth factor-β1 in human colorectal cancer correlates with reduced circulating dendritic cells and increased colonic langerhans cell infiltration. Clin. Exp. Immunol. 2003, 134, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.G.; Lysaght, J.; Todryk, S.; Mills, K.H. Suppression of antitumor immunity by IL-10 and TGF-β-producing T cells infiltrating the growing tumor: Influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Inge, T.H.; Hoover, S.K.; Susskind, B.M.; Barrett, S.K.; Bear, H.D. Inhibition of tumor-specific cytotoxic T-lymphocyte responses by transforming growth factor β 1. Cancer Res. 1992, 52, 1386–1392. [Google Scholar] [PubMed]
- Zhang, M.; Berndt, B.E.; Chen, J.J.; Kao, J.Y. Expression of a soluble TGF-β receptor by tumor cells enhances dendritic cell/tumor fusion vaccine efficacy. J. Immunol. 2008, 181, 3690–3697. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Kan, S.; Takakura, K.; Kajihara, M.; Uchiyama, K.; Hara, E.; Ohkusa, T.; et al. Improved immunogenicity of fusions between ethanol-treated cancer cells and dendritic cells exposed to dual TLR stimulation. Oncoimmunology 2013, 2, e25375. [Google Scholar] [CrossRef] [PubMed]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Copier, J.; Dalgleish, A. Whole-cell vaccines: A failure or a success waiting to happen? Curr. Opin. Mol. Ther. 2010, 12, 14–20. [Google Scholar] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Takahara, A.; Odahara, S.; Tsukinaga, S.; Yukawa, T.; Mitobe, J.; et al. Augmentation of antitumor immunity by fusions of ethanol-treated tumor cells and dendritic cells stimulated via Dual TLRs through TGF-β1 blockade and IL-12p70 production. PLoS ONE 2013, 8, e63498. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Strategies to improve the immunogenicity of anticancer vaccines based on dendritic cell/malignant cell fusions. Oncoimmunology 2013, 2, e25994. [Google Scholar] [CrossRef]
- Hirohashi, Y.; Torigoe, T.; Inoda, S.; Takahashi, A.; Morita, R.; Nishizawa, S.; Tamura, Y.; Suzuki, H.; Toyota, M.; Sato, N. Immune response against tumor antigens expressed on human cancer stem-like cells/tumor-initiating cells. Immunotherapy 2010, 2, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Li, F.; Luo, B.; Wang, X.H.; Sun, H.C.; Liu, S.; Cui, Y.Q.; Xu, X.X. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma 2009, 56, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Hirohashi, Y.; Torigoe, T.; Tsukahara, T.; Kanaseki, T.; Kochin, V.; Sato, N. Immune responses to human cancer stem-like cells/cancer-initiating cells. Cancer Sci. 2016, 107, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A National Cancer Institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Qin, K.; Tian, G.; Li, P.; Chen, Q.; Zhang, R.; Ke, Y.Q.; Xiao, Z.C.; Jiang, X.D. Anti-glioma response of autologous T cells stimulated by autologous dendritic cells electrofused with CD133+ or CD133− glioma cells. J. Neuroimmunol. 2012, 242, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Engelmann, K.; Shen, H.; Finn, O.J. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008, 68, 2419–2426. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Gong, J.; Calderwood, S.K. Purification, preparation, and use of chaperone-peptide complexes for tumor immunotherapy. Methods Mol. Biol. 2013, 960, 209–217. [Google Scholar] [PubMed]
- Murshid, A.; Gong, J.; Calderwood, S.K. Hsp90-peptide complexes stimulate antigen presentation through the class II pathway after binding scavenger receptor SREC-I. Immunobiology 2014, 219, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Georgopoulos, C.; Welch, W.J. Role of the major heat shock proteins as molecular chaperones. Annu. Rev. Cell Biol. 1993, 9, 601–634. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, Y.; Bharti, A.; Khaleque, A.A.; Song, B.; Liu, C.; Apostolopoulos, V.; Xing, P.X.; Calderwood, S.K.; Gong, J. Enhanced immunogenicity of heat shock protein 70 peptide complexes from dendritic cell-tumor fusion cells. J. Immunol. 2006, 177, 5946–5955. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Zhang, Y.; Durfee, J.; Weng, D.; Liu, C.; Koido, S.; Song, B.; Apostolopoulos, V.; Calderwood, S.K. A heat shock protein 70-based vaccine with enhanced immunogenicity for clinical use. J. Immunol. 2010, 184, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, W.; Wang, Y.; Chen, J.; Liu, Y.; Zhang, Y. Enhanced antitumor immunity of nanoliposome-encapsulated heat shock protein 70 peptide complex derived from dendritic tumor fusion cells. Oncol. Rep. 2015, 33, 2695–2702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Chen, J.; Liu, Y.; Luo, W. Dendritic-tumor fusion cells derived heat shock protein70-Peptide complex has enhanced immunogenicity. PLoS ONE 2015, 10, e0126075. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L.; Tesniere, A.; Aymeric, L.; Ma, Y.; Ortiz, C.; Vermaelen, K.; Panaretakis, T.; Mignot, G.; Ullrich, E.; et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat. Med. 2009, 15, 1170–1178. [Google Scholar] [CrossRef] [PubMed]
- Adkins, I.; Fucikova, J.; Garg, A.D.; Agostinis, P.; Špíšek, R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology 2014, 3, e968434. [Google Scholar] [CrossRef] [PubMed]
- Gameiro, S.R.; Jammeh, M.L.; Wattenberg, M.M.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget 2014, 5, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Schmitt, E.; Hamai, A.; Hervas-Stubbs, S.; Obeid, M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Combination of chemotherapy and immunotherapy for cancer: A paradigm revisited. Lancet Oncol. 2007, 8, 2–3. [Google Scholar] [CrossRef]
- Lesterhuis, W.J.; Haanen, J.B.; Punt, C.J. Cancer immunotherapy—Revisited. Nat. Rev. Drug Discov. 2011, 10, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Ito, A.; Shinkai, M.; Honda, H.; Yoshikawa, K.; Saga, S.; Wakabayashi, T.; Yoshida, J.; Kobayashi, T. Heat shock protein 70 expression induces antitumor immunity during intracellular hyperthermia using magnetite nanoparticles. Cancer Immunol. Immunother. 2003, 52, 80–88. [Google Scholar] [PubMed]
- Apetoh, L.; Ghiringhelli, F.; Tesniere, A.; Obeid, M.; Ortiz, C.; Criollo, A.; Mignot, G.; Maiuri, M.C.; Ullrich, E.; Saulnier, P.; et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 2007, 13, 1050–1059. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Kan, S.; Yoshida, K.; Yoshizaki, S.; Takakura, K.; Namiki, Y.; Tsukinaga, S.; Odahara, S.; Kajihara, M.; Okamoto, M.; et al. Immunogenic modulation of cholangiocarcinoma cells by chemoimmunotherapy. Anticancer Res. 2014, 34, 6353–6361. [Google Scholar] [PubMed]
- Takahara, A.; Koido, S.; Ito, M.; Nagasaki, E.; Sagawa, Y.; Iwamoto, T.; Komita, H.; Ochi, T.; Fujiwara, H.; Yasukawa, M.; et al. Gemcitabine enhances Wilms' tumor gene WT1 expression and sensitizes human pancreatic cancer cells with WT1-specific T-cell-mediated antitumor immune response. Cancer Immunol. Immunother. 2011, 60, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Koido, S.; Okamoto, M.; Hayashi, K.; Ito, M.; Kamata, Y.; Komita, H.; Ishidao, T.; Nagasaki, E.; Homma, S. Gemcitabine treatment enhances HER2 expression in low HER2-expressing breast cancer cells and enhances the antitumor effects of trastuzumab emtansine. Oncol. Rep. 2015, 34, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Koido, S.; Okamoto, M.; Hayashi, K.; Ito, M.; Kamata, Y.; Komita, H.; Nagasaki, E.; Homma, S. Up-regulation of HER2 by gemcitabine enhances the antitumor effect of combined gemcitabine and trastuzumab emtansine treatment on pancreatic ductal adenocarcinoma cells. BMC Cancer 2015, 15, 726. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Hara, E.; Mitsunaga, M.; Namiki, Y.; Takahara, A.; Nagasaki, E.; Komita, H.; Sagawa, Y.; Ohkusa, T.; et al. In vitro generation of cytotoxic and regulatory T cells by fusions of human dendritic cells and hepatocellular carcinoma cells. J. Transl. Med. 2008, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kan, S.; Hazama, S.; Maeda, K.; Inoue, Y.; Homma, S.; Koido, S.; Okamoto, M.; Oka, M. Suppressive effects of cyclophosphamide and gemcitabine on regulatory T-cell induction in vitro. Anticancer Res. 2012, 32, 5363–5369. [Google Scholar] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Vasir, B.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Reddy, V.; Dannull, J.; Ding, E.; Nair, S.K.; Tyler, D.S.; Pruitt, S.K.; Lee, W.T. Impact of anti-CD25 monoclonal antibody on dendritic cell-tumor fusion vaccine efficacy in a murine melanoma model. J. Transl. Med. 2013, 11, 148. [Google Scholar] [CrossRef] [PubMed]
- Avigan, D.; Rosenblatt, J.; Kufe, D. Dendritic/tumor fusion cells as cancer vaccines. Semin. Oncol. 2012, 39, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Advantages | DC-tumor FCs present whole tumor-derived antigenic peptides, which avoids the need to identify antigenic peptides for individual patients. |
| A broad array of known and unidentified tumor-associated antigens are simultaneously presented on the surface of DC-tumor FCs. | |
| Endogenously-synthesized tumor-associated antigens in DC-tumor FCs are better access to MHC class I and II molecules. | |
| Increased the frequency of polyclonal antigen-specific CD4+ and CD8+ T cells can be induced by DC-tumor FCs. | |
| DC-tumor FCs can induce long-term efficient antitumor immunity. | |
| Numerous tumor-associated antigens are presented in the context of co-stimulatory molecules in DC-tumor FCs. | |
| DC-tumor FCs prevent tolerance induction. | |
| Autologous DC-autologous tumor FCs do not have to take up exogenous TAAs in order to activate CD4+ and CD8+ T cells. | |
| Modifications of DCs and tumor cells are independently possible while their characters present after the fusion. | |
| Allogeneic DC and allogeneic tumor cells can be used instead of autologous cells in generation of DC-tumor FCs. | |
| DC-tumor FC-based cancer vaccines can be combined with standard therapies. | |
| Disadvantages | The limited availability of viable autologous tumor cells as a fusion partner. |
| Induction of antigen-specific CD4+ and CD8+ T cell responses by allogeneic DC-tumor FCs are at least partly associated with sharing of MHC class I. | |
| Fusion efficiency depends on cell conditions due to the sensitivity of cells to PEG treatment. |
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koido, S. Dendritic-Tumor Fusion Cell-Based Cancer Vaccines. Int. J. Mol. Sci. 2016, 17, 828. https://doi.org/10.3390/ijms17060828
Koido S. Dendritic-Tumor Fusion Cell-Based Cancer Vaccines. International Journal of Molecular Sciences. 2016; 17(6):828. https://doi.org/10.3390/ijms17060828
Chicago/Turabian StyleKoido, Shigeo. 2016. "Dendritic-Tumor Fusion Cell-Based Cancer Vaccines" International Journal of Molecular Sciences 17, no. 6: 828. https://doi.org/10.3390/ijms17060828
APA StyleKoido, S. (2016). Dendritic-Tumor Fusion Cell-Based Cancer Vaccines. International Journal of Molecular Sciences, 17(6), 828. https://doi.org/10.3390/ijms17060828