
Could Vitamin D Analogues Be Used to Target Leukemia Stem Cells?

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. The Modern Approach in Cancer Therapy
3. A New Approach Aims to Target Cancer Stem Cell (CSCs)
4. Practical Implications of the Leukemic Stem Cells (LSCs) in Therapy
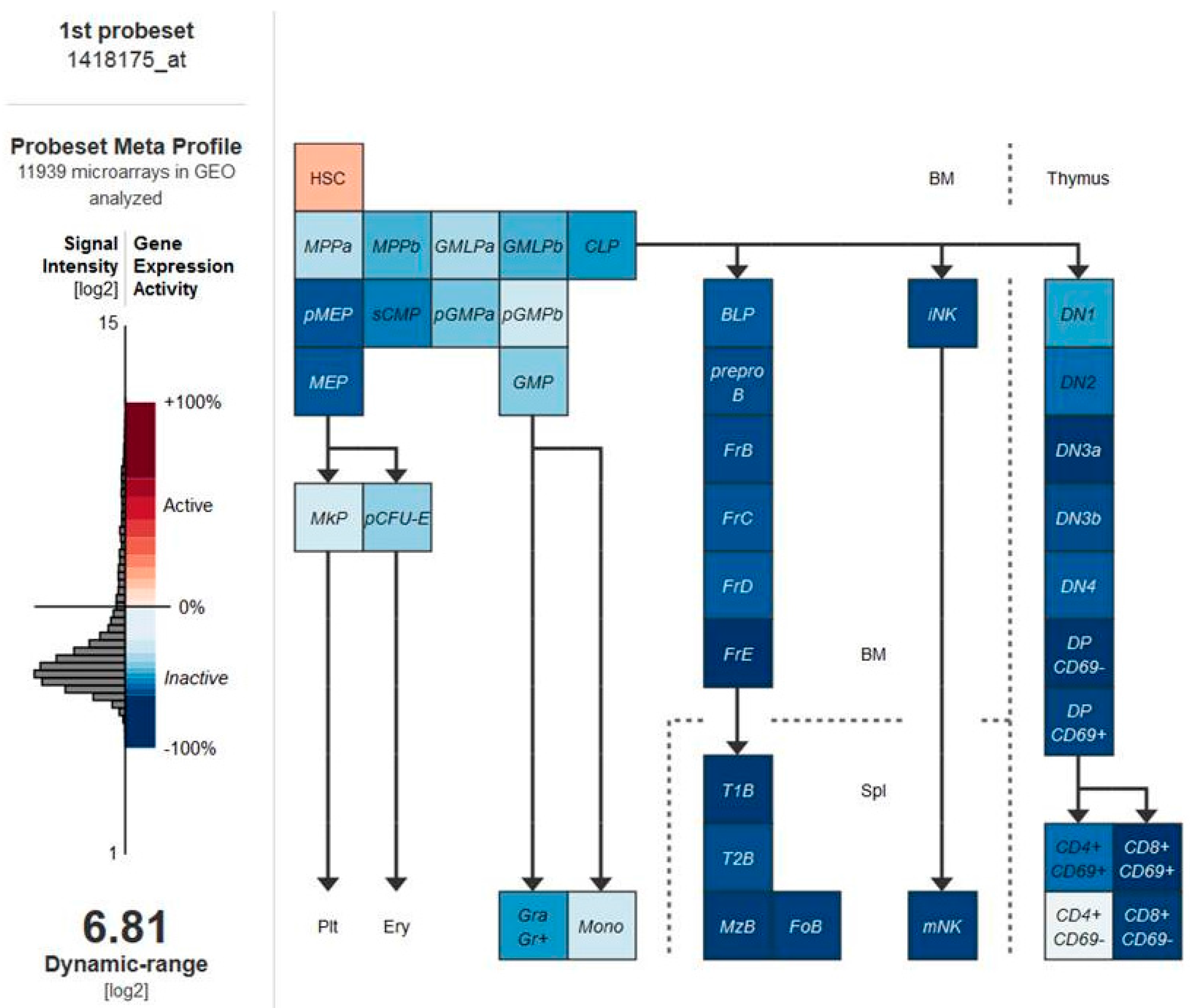
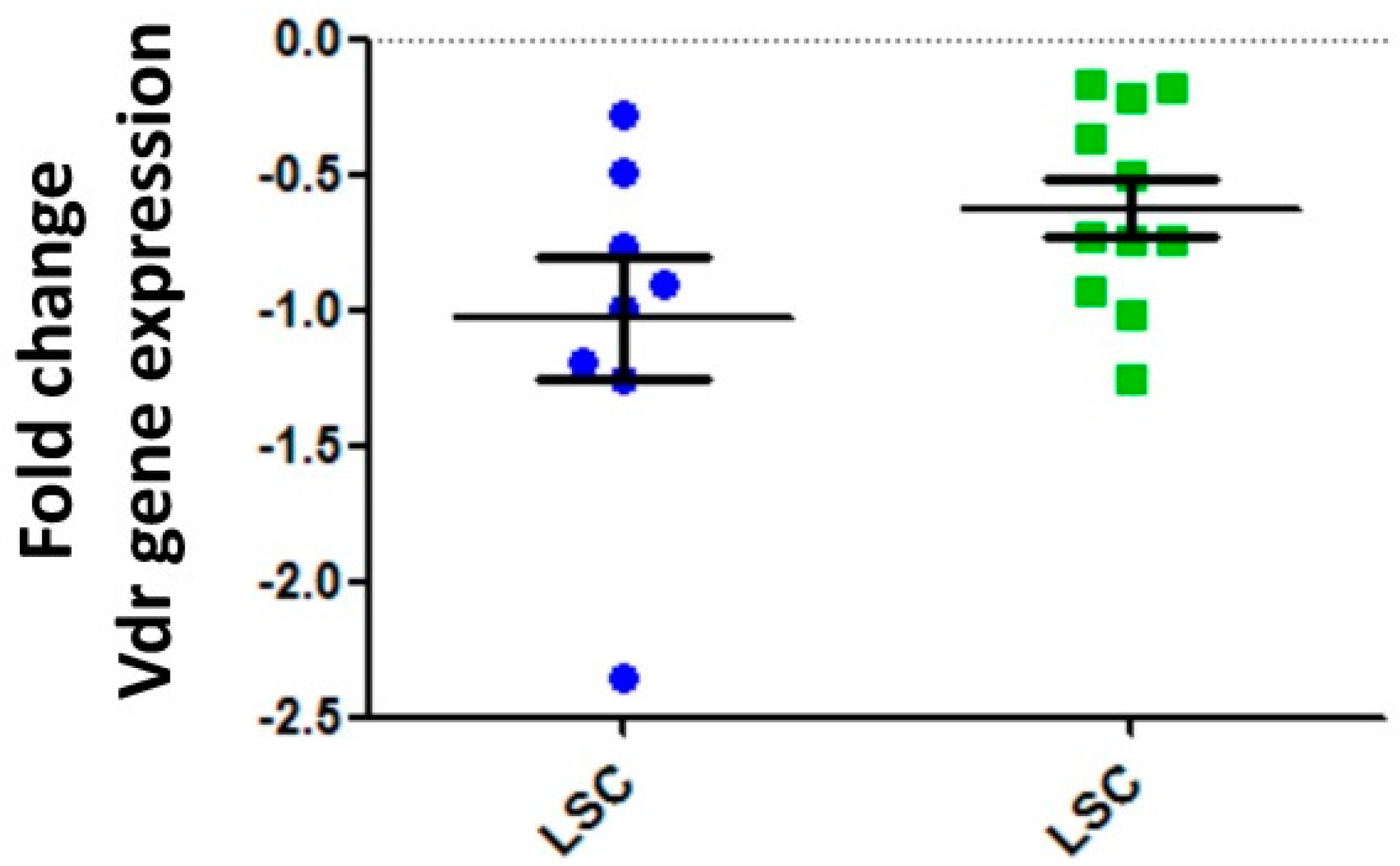
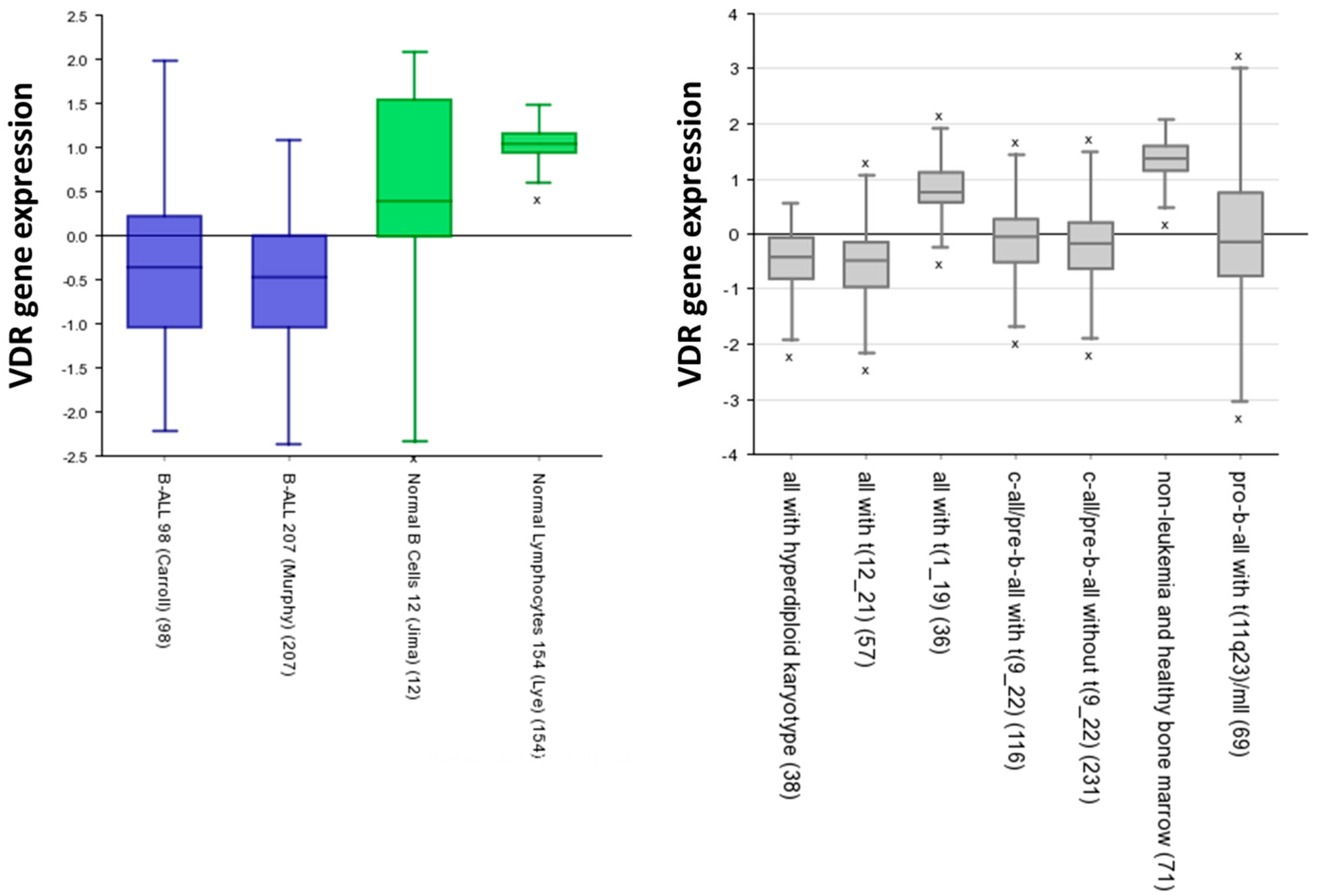
5. Vitamin D Receptors (VDRs) on Hematopoietic Cells and LSCs
6. Vitamin D and the Epigenome
7. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Etzioni, R.; Urban, N.; Ramsey, S.; McIntosh, M.; Schwartz, S.; Reid, B.; Radich, J.; Anderson, G.; Hartwell, L. The case for early detection. Nat. Rev. Cancer 2003, 3, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caro, M.; Sanchez-Garcia, I. Killing time for cancer stem cells (CSC): Discovery and development of selective CSC inhibitors. Curr. Med. Chem. 2006, 13, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A.; Roberts, T.G., Jr. Timeline: Chemotherapy and the war on cancer. Nat. Rev. Cancer 2005, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Hope, K.J.; Jin, L.; Dick, J.E. Acute myeloid leukemia originates from a hierarchy of leukemic stem cell classes that differ in self-renewal capacity. Nat. Immunol. 2004, 5, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Weissman, I.L.; Akashi, K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8;21 chromosomal translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 7521–7526. [Google Scholar] [CrossRef] [PubMed]
- Cobaleda, C.; Gutierrez-Cianca, N.; Perez-Losada, J.; Flores, T.; Garcia-Sanz, R.; Gonzalez, M.; Sanchez-Garcia, I. A primitive hematopoietic cell is the target for the leukemic transformation in human philadelphia-positive acute lymphoblastic leukemia. Blood 2000, 95, 1007–1013. [Google Scholar] [PubMed]
- Cox, C.V.; Evely, R.S.; Oakhill, A.; Pamphilon, D.H.; Goulden, N.J.; Blair, A. Characterization of acute lymphoblastic leukemia progenitor cells. Blood 2004, 104, 2919–2925. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.V.; Martin, H.M.; Kearns, P.R.; Virgo, P.; Evely, R.S.; Blair, A. Characterization of a progenitor cell population in childhood T-cell acute lymphoblastic leukemia. Blood 2007, 109, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Ren, R. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer 2005, 5, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.V.; Barnes, D.J. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat. Rev. Cancer 2007, 7, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Dick, J.E. Breast cancer stem cells revealed. Proc. Natl. Acad. Sci. USA 2003, 100, 3547–3549. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.F.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Cobaleda, C.; Sanchez-Garcia, I. B-cell acute lymphoblastic leukaemia: Towards understanding its cellular origin. Bioessays 2009, 31, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.; Sanchez-Garcia, I. Is lineage decision-making restricted during tumoral reprograming of haematopoietic stem cells? Oncotarget 2015, 6, 43326–43341. [Google Scholar] [PubMed]
- Sanchez-Garcia, I. Getting to the stem of cancer. Semin Cancer Biol. 2010, 20, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Hauer, J.; Ruiz-Roca, L.; Ingenhag, D.; Rodriguez-Meira, A.; Auer, F.; Borkhardt, A.; Sanchez-Garcia, I. Tumoral stem cell reprogramming as a driver of cancer: Theory, biological models, implications in cancer therapy. Semin. Cancer Biol. 2015, 32, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garcia, I. How tumour cell identity is established? Semin. Cancer Biol. 2015, 32, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Ries, L.A.G.; Harkins, D.; Krapcho, M.; Mariotto, A.; Miller, B.; Feuer, E.J.; Clegg, L.; Eisner, M.P.; Horner, M.J.; Howlader, N.; et al. Seer Cancer Statistics Review, 1975–2003. National Cancer Institute: Bethesda, MD, USA. Available online: http://seer.Cancer.Gov/csr/1975_2003/ (accessed on 30 March 2016).
- Cohnheim, J. Ueber entzundung und eiterung. Pathol. Anat. Physiol. Klin. Med. 1867, 40, 1–79. [Google Scholar] [CrossRef]
- Virchow, R. (Ed.) Virchows Arch. Pathol. Anat. Physiol. Klin. Med. 1855, 3, 23.
- Vicente-Duenas, C.; Romero-Camarero, I.; Cobaleda, C.; Sanchez-Garcia, I. Function of oncogenes in cancer development: A changing paradigm. EMBO J. 2013, 32, 1502–1513. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lorenzo, A.; Gonzalez-Herrero, I.; Rodriguez-Hernandez, G.; Garcia-Ramirez, I.; Vicente-Duenas, C.; Sanchez-Garcia, I. Early epigenetic cancer decisions. Biol. Chem. 2014, 395, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caro, M.; Cobaleda, C.; Gonzalez-Herrero, I.; Vicente-Duenas, C.; Bermejo-Rodriguez, C.; Sanchez-Beato, M.; Orfao, A.; Pintado, B.; Flores, T.; Sanchez-Martin, M.; et al. Cancer induction by restriction of oncogene expression to the stem cell compartment. EMBO J. 2009, 28, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Duenas, C.; Perez-Caro, M.; Abollo-Jimenez, F.; Cobaleda, C.; Sanchez-Garcia, I. Stem-cell driven cancer: “Hands-off” regulation of cancer development. Cell Cycle 2009, 8, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicente-Duenas, C.; Gonzalez-Herrero, I.; Cenador, M.B.; Criado, F.J.; Sanchez-Garcia, I. Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by mafb. Cell Cycle 2012, 11, 3896–3900. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Romero-Camarero, I.; Gonzalez-Herrero, I.; Alonso-Escudero, E.; Abollo-Jimenez, F.; Jiang, X.; Gutierrez, N.C.; Orfao, A.; Marin, N.; Villar, L.M.; et al. A novel molecular mechanism involved in multiple myeloma development revealed by targeting mafb to haematopoietic progenitors. EMBO J. 2012, 31, 3704–3717. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Duenas, C.; Fontan, L.; Gonzalez-Herrero, I.; Romero-Camarero, I.; Segura, V.; Aznar, M.A.; Alonso-Escudero, E.; Campos-Sanchez, E.; Ruiz-Roca, L.; Barajas-Diego, M.; et al. Expression of MALT1 oncogene in hematopoietic stem/progenitor cells recapitulates the pathogenesis of human lymphoma in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 10534–10539. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Hernandez, T.; Vicente-Duenas, C.; Sanchez-Garcia, I.; Martin-Zanca, D. P53 restoration kills primitive leukemia cells in vivo and increases survival of leukemic mice. Cell Cycle 2012, 12, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Romero-Camarero, I.; Jiang, X.; Natkunam, Y.; Lu, X.; Vicente-Duenas, C.; Gonzalez-Herrero, I.; Flores, T.; Luis Garcia, J.; McNamara, G.; Kunder, C.; et al. Germinal centre protein HGAL promotes lymphoid hyperplasia and amyloidosis via BCR-mediated Syk activation. Nat. Commun. 2013, 4, 1338. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Vicente-Duenas, C.; Romero-Camarero, I.; Long Liu, C.; Dai, B.; Gonzalez-Herrero, I.; Garcia-Ramirez, I.; Alonso-Escudero, E.; Iqbal, J.; Chan, W.C.; et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat. Commun. 2014, 5, 3904. [Google Scholar] [CrossRef] [PubMed]
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARγ agonists. Nature 2015, 525, 380–383. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.C.; Juckett, M.B. The role of vitamin D in hematologic disease and stem cell transplantation. Nutrients 2013, 5, 2206–2221. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Sahoo, D.; Rossi, D.J.; Bhattacharya, D.; Serwold, T.; Inlay, M.A.; Ehrlich, L.I.; Fathman, J.W.; Dill, D.L.; Weissman, I.L. Gene expression commons: An open platform for absolute gene expression profiling. PLoS ONE 2012, 7, e40321. [Google Scholar] [CrossRef] [PubMed]
- R2: Genomics analysis and visualization platform. Available online: http://r2.Amc.nl (accessed on 30 March 2016).
- Martin-Lorenzo, A.; Hauer, J.; Vicente-Duenas, C.; Auer, F.; Gonzalez-Herrero, I.; Garcia-Ramirez, I.; Ginzel, S.; Thiele, R.; Constantinescu, S.N.; Bartenhagen, C.; et al. Infection exposure is a causal factor in b-cell precursor acute lymphoblastic leukemia as a result of pax5-inherited susceptibility. Cancer Discov. 2015, 5, 1328–1343. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Varga, F. Impact of vitamin D metabolism on clinical epigenetics. Clin. Epigenet. 2011, 2, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, X.; Shi, H.; Su, S.; Harshfield, G.A.; Gutin, B.; Snieder, H.; Dong, Y. A genome-wide methylation study of severe vitamin D deficiency in african american adolescents. J. Pediatr. 2013, 162, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.J.; Marcinkowska, E.; Gocek, E.; Studzinski, G.P.; Brown, G. Vitamin D3-driven signals for myeloid cell differentiation--Implications for differentiation therapy. Leuk. Res. 2010, 34, 553–565. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, A.V.; Trump, D.L.; Johnson, C.S.; Feldman, D. The role of vitamin D in cancer prevention and treatment. Rheum. Dis. Clin. N. Am. 2012, 38, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Smigiel, J.M.; Junk, D.J.; Jackson, M.W. Cancer stem cell plasticity drives therapeutic resistance. Cancers (Basel) 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Meacham, C.E.; Morrison, S.J. Tumour heterogeneity and cancer cell plasticity. Nature 2013, 501, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Ramírez, I.; Martín-Lorenzo, A.; González-Herrero, I.; Rodriguez-Hernández, G.; Vicente-Dueñas, C.; Sánchez-García, I. Could Vitamin D Analogues Be Used to Target Leukemia Stem Cells? Int. J. Mol. Sci. 2016, 17, 889. https://doi.org/10.3390/ijms17060889
García-Ramírez I, Martín-Lorenzo A, González-Herrero I, Rodriguez-Hernández G, Vicente-Dueñas C, Sánchez-García I. Could Vitamin D Analogues Be Used to Target Leukemia Stem Cells? International Journal of Molecular Sciences. 2016; 17(6):889. https://doi.org/10.3390/ijms17060889
Chicago/Turabian StyleGarcía-Ramírez, Idoia, Alberto Martín-Lorenzo, Inés González-Herrero, Guillermo Rodriguez-Hernández, Carolina Vicente-Dueñas, and Isidro Sánchez-García. 2016. "Could Vitamin D Analogues Be Used to Target Leukemia Stem Cells?" International Journal of Molecular Sciences 17, no. 6: 889. https://doi.org/10.3390/ijms17060889
APA StyleGarcía-Ramírez, I., Martín-Lorenzo, A., González-Herrero, I., Rodriguez-Hernández, G., Vicente-Dueñas, C., & Sánchez-García, I. (2016). Could Vitamin D Analogues Be Used to Target Leukemia Stem Cells? International Journal of Molecular Sciences, 17(6), 889. https://doi.org/10.3390/ijms17060889