
Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
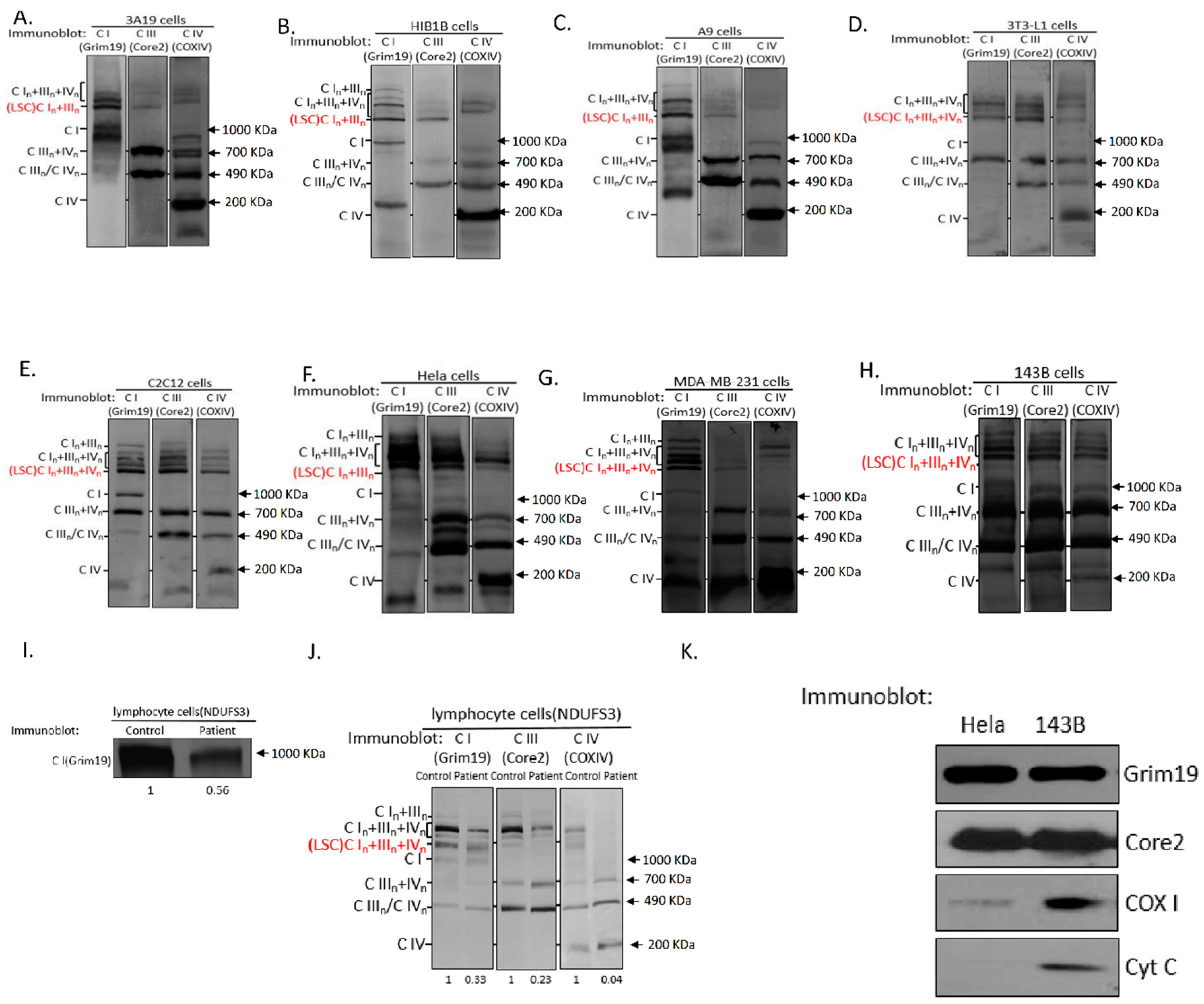
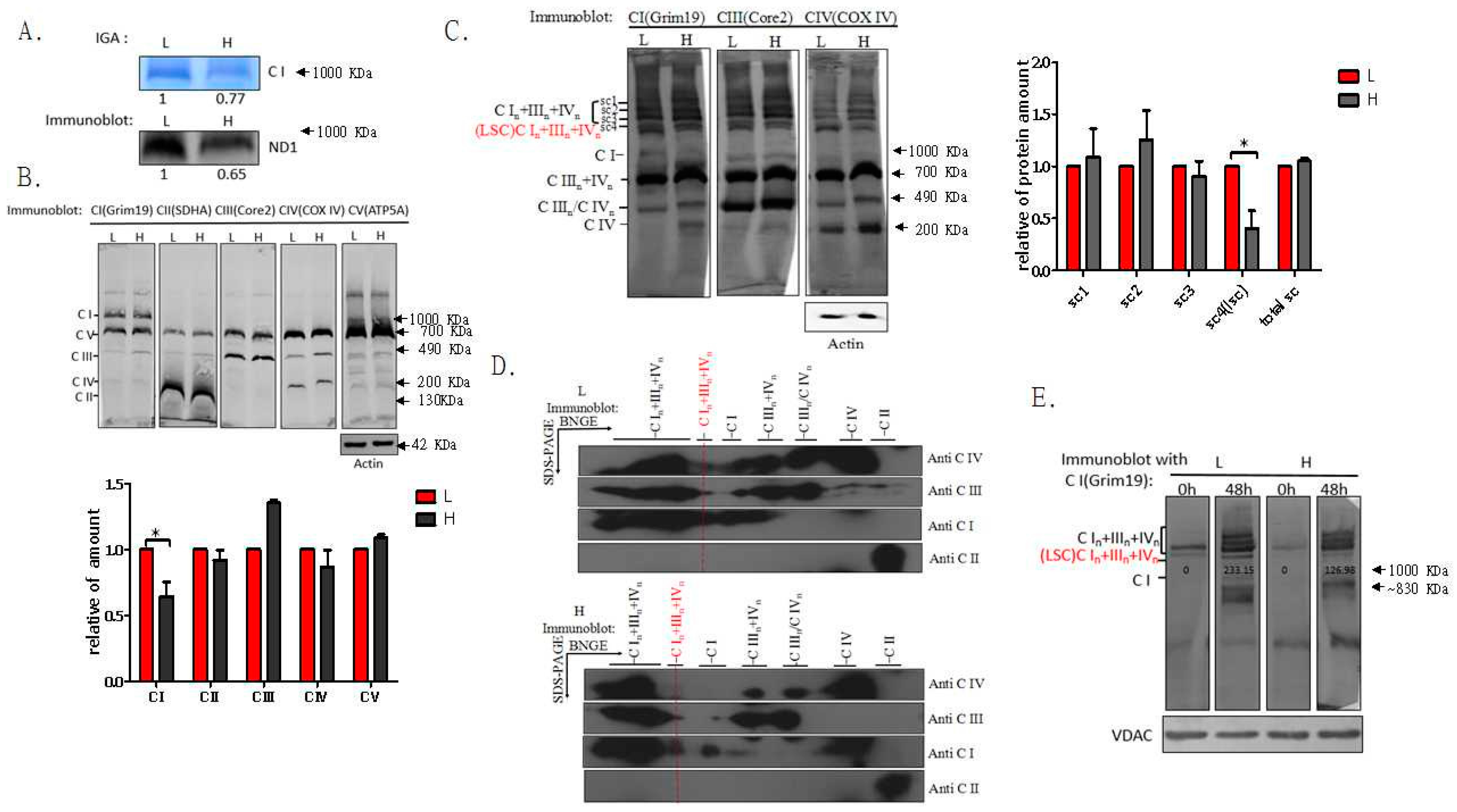
2.1. Respiratory Chain Supercomplexes in Humans and Mice
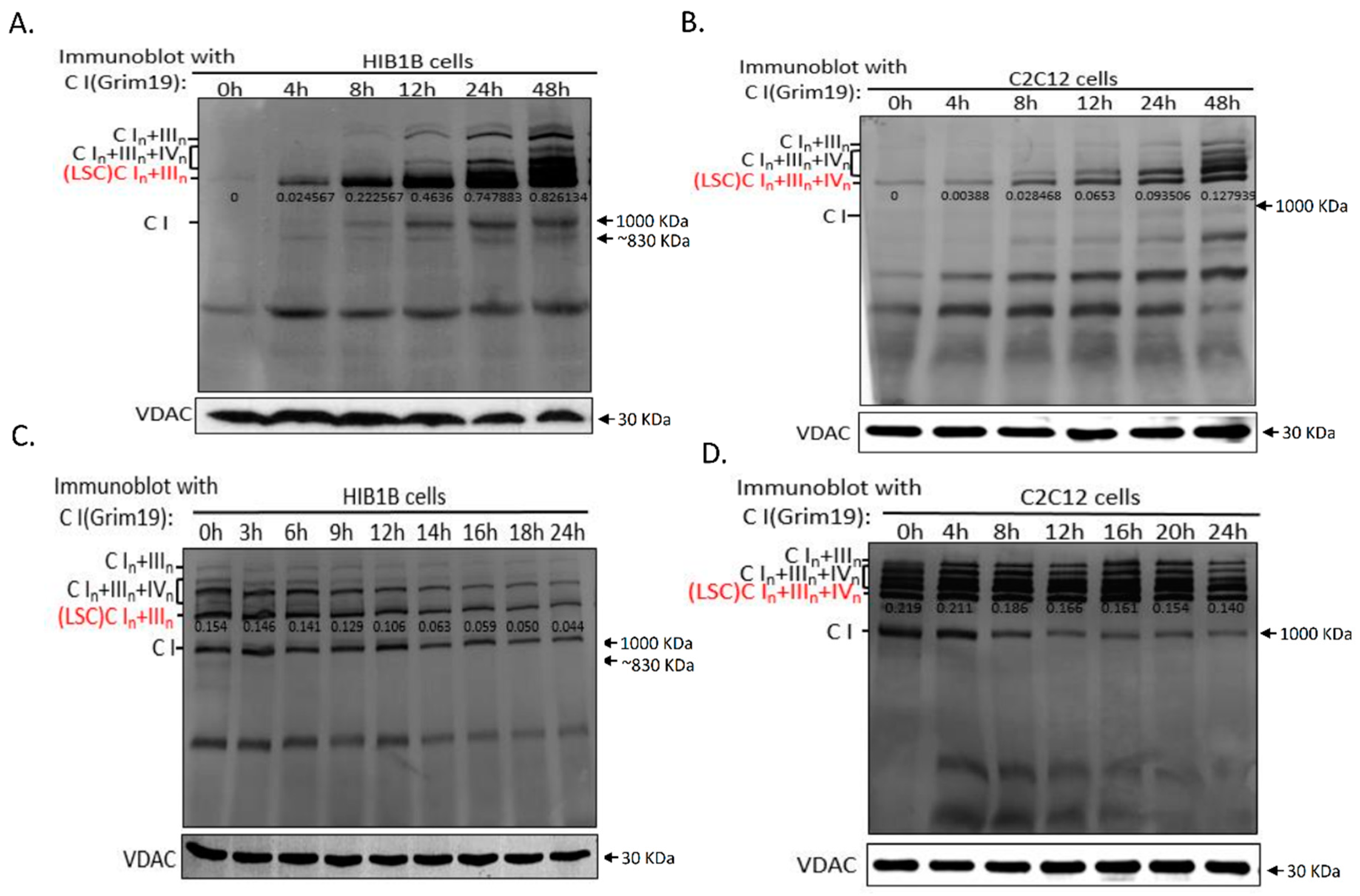
2.2. Dynamics of Respiratory Chain LSC
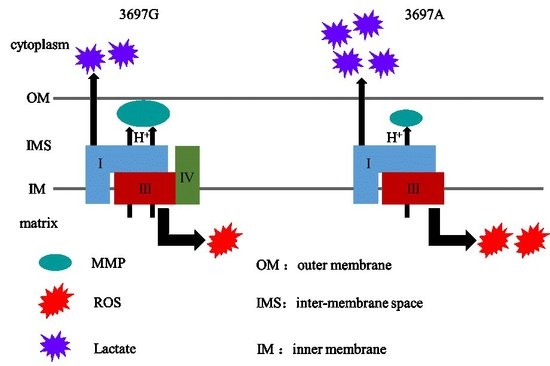
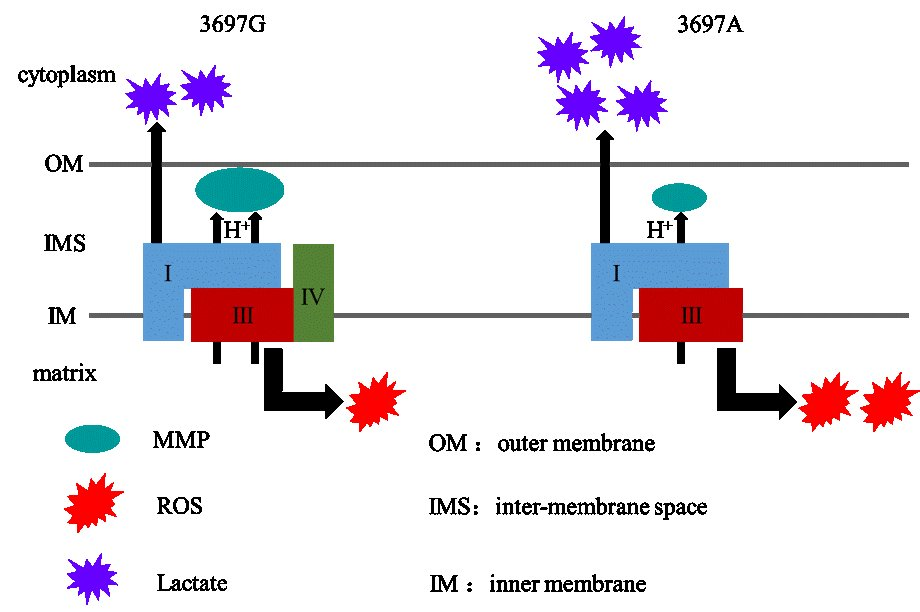
2.3. Mitochondrial NADH Dehydrogenase Subunit 1 (MT-ND1) 3697G>A Mutation Impairs LSC Formation
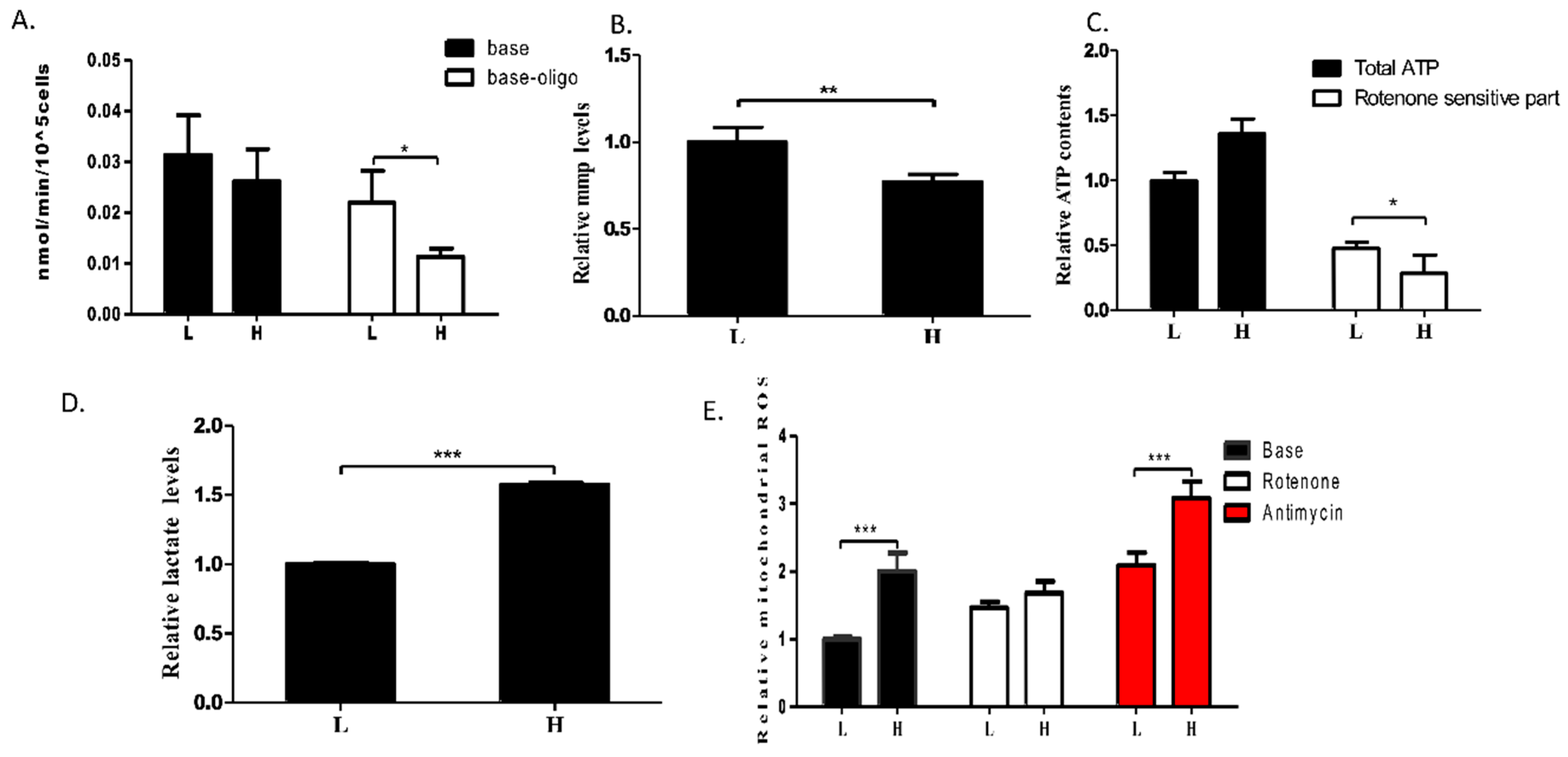
The MT-ND1 3697G>A Mutation Induces OXPHOS Defect
3. Discussion
4. Materials and Methods
4.1. Characterization of Patients
4.2. Generation of Cell Lines and Culture Conditions
4.3. DNA Analysis
4.4. mtDNA Copy Number Quantification
4.5. Mitochondria Isolation from Mouse Liver and Cell Lines, Protein Preparation, Blue Native PAGE, and in-Gel Activity Assay
4.6. Antibodies and Immunoblotting
4.7. Lactate Measurement
4.8. ROS Measurement
4.9. MMP and ATP Measurements
4.10. Oxygen Consumption
4.11. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BN-PAGE | blue native polyacrylamide gel electrophoresis |
| ATP | adenosine triphosphate |
| ADP | adenosine diphosphate |
| mtDNA | mitochondrial DNA |
| MMP | mitochondrial membrane potential |
| ROS | reactive oxygen species |
| OXPHOS | oxidative phosphorylation system |
| cyt c | cytochrome c |
| ETC | electron transfer chain |
| NDUFS3 | NADH dehydrogenase (ubiquinone) iron-sulfur protein 3 |
| CAP | chloramphenicol |
| MELAS | mitochondrial encephalopathy, lactic acidosis, and stroke-like episode |
| NDUFA13/Grim19 | NADH dehydrogenase (ubiquinone) 1 α subcomplex, 13 |
| ND1 | NADH dehydrogenase subunit 1 |
| SCAFs | supercomplex assembly factors |
| Rcf1/2 | respiratory supercomplex factors 1 and 2 |
| COX7a21 | cyt c oxidase subunit 7A-related protein |
| DDM | N-dodecyl-β-d-maltoside |
| NBT | nitrotetrazolium blue chloride |
| DAB | 3,3’-diamiobenzidine tetra hydrochloride hydrate |
| BCIP | 5-bromo-4-chloro-3’-indolyphosphate p-toluidine |
| TMRM | Tetramethylrhodamine methyl ester |
References
- Wallace, D.C. A mitochondrial bioenergetic etiology of disease. J. Clin. Investig. 2013, 123, 1405–1412. [Google Scholar] [CrossRef] [PubMed]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. A simple and rapid assay of oxidative phosphorylation. Nature 1955, 175, 1120–1121. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta 2014, 1837, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Hornig-Do, H.T.; Tatsuta, T.; Buckermann, A.; Bust, M.; Kollberg, G.; Rotig, A.; Hellmich, M.; Nijtmans, L.; Wiesner, R.J. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 2012, 31, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; D’Aurelio, M.; Deng, J.H.; Park, J.S.; Manfredi, G.; Hu, P.; Lu, J.; Bai, Y. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 2007, 282, 17557–17562. [Google Scholar] [CrossRef] [PubMed]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Blaza, J.N.; Serreli, R.; Jones, A.J.; Mohammed, K.; Hirst, J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. USA 2014, 111, 15735–15740. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H. Respiratory chain supercomplexes. IUBMB Life 2001, 52, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Althoff, T.; Mills, D.J.; Popot, J.L.; Kuhlbrandt, W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef] [PubMed]
- Dudkina, N.V.; Kudryashev, M.; Stahlberg, H.; Boekema, E.J. Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2011, 108, 15196–15200. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Mourier, A.; Matic, S.; Ruzzenente, B.; Larsson, N.G.; Milenkovic, D. The respiratory chain supercomplex organization is independent of COX7a2l isoforms. Cell Metab. 2014, 20, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Quiros, P.M.; Espanol, Y.; Acin-Perez, R.; Rodriguez, F.; Barcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernandez-Garcia, M.S.; Fueyo, A.; et al. ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [PubMed]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res. 2008, 80, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Yaffe, D.; Saxel, O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977, 270, 725–727. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Attardi, G. The mtDNA-encoded ND6 subunit of mitochondrial NADH dehydrogenase is essential for the assembly of the membrane arm and the respiratory function of the enzyme. EMBO J. 1998, 17, 4848–4858. [Google Scholar] [CrossRef] [PubMed]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.R.; Choy, L.; Graves, R.A.; Fox, N.; Solevjeva, V.; Klaus, S.; Ricquier, D.; Spiegelman, B.M. Hibernoma formation in transgenic mice and isolation of a brown adipocyte cell line expressing the uncoupling protein gene. Proc. Natl. Acad. Sci. USA 1992, 89, 7561–7565. [Google Scholar] [CrossRef] [PubMed]
- Shinzawa-Itoh, K.; Shimomura, H.; Yanagisawa, S.; Shimada, S.; Takahashi, R.; Oosaki, M.; Ogura, T.; Tsukihara, T. Purification of active respiratory supercomplex from bovine heart mitochondria enables functional studies. J. Biol. Chem. 2016, 291, 4178–4184. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Kirby, D.M.; McFarland, R.; Ohtake, A.; Dunning, C.; Ryan, M.T.; Wilson, C.; Ketteridge, D.; Turnbull, D.M.; Thorburn, D.R.; Taylor, R.W. Mutations of the mitochondrial ND1 gene as a cause of MELAS. J. Med. Genet. 2004, 41, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Negishi, Y.; Hattori, A.; Takeshita, E.; Sakai, C.; Ando, N.; Ito, T.; Goto, Y.-I.; Saitoh, S. Homoplasmy of a mitochondrial 3697G>A mutation causes Leigh syndrome. J. Hum. Genet. 2014, 59, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Schägger, H.; Pfeiffer, K. The ratio of oxidative phosphorylation complexes I–V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 2001, 276, 37861–37867. [Google Scholar] [PubMed]
- Spangenberg, L.; Grana, M.; Greif, G.; Suarez-Rivero, J.M.; Krysztal, K.; Tapie, A.; Boidi, M.; Fraga, V.; Lemes, A.; Guecaimburu, R.; et al. 3697G>A in MT-ND1 is a causative mutation in mitochondrial disease. Mitochondrion 2016, 28, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Valente, L.; Piga, D.; Lamantea, E.; Carrara, F.; Uziel, G.; Cudia, P.; Zani, A.; Farina, L.; Morandi, L.; Mora, M.; et al. Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta 2009, 1787, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Shiba, S.; Horie-Inoue, K.; Shimokata, K.; Inoue, S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Taylor, E.B.; Dephoure, N.; Heo, J.M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vogtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 mediates cytochrome oxidase assembly and respirasome formation, revealing heterogeneity of the enzyme complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Baracca, A.; Barbero, G.; Bergamini, C.; Dalmonte, M.E.; del Sole, M.; Faccioli, M.; Falasca, A.; Fato, R.; Genova, M.L.; et al. Mitochondrial respiratory chain super-complex I-III in physiology and pathology. Biochim. Biophys. Acta 2010, 1797, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Dudkina, N.V.; Jansch, L.; Braun, H.-P.; Boekema, E.J. A structural investigation of complex I and I + III2 supercomplex from Zea mays at 11–13 A resolution: Assignment of the carbonic anhydrase domain and evidence for structural heterogeneity within complex I. Biochim. Biophys. Acta 2008, 1777, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Schafer, E.; Seelert, H.; Reifschneider, N.H.; Krause, F.; Dencher, N.A.; Vonck, J. Architecture of active mammalian respiratory chain supercomplexes. J. Biol. Chem. 2006, 281, 15370–15375. [Google Scholar] [CrossRef] [PubMed]
- Heide, H.; Bleier, L.; Steger, M.; Ackermann, J.; Drose, S.; Schwamb, B.; Zornig, M.; Reichert, A.S.; Koch, I.; Wittig, I.; et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012, 16, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Chomyn, A.; Lai, S.T.; Shakeley, R.; Bresolin, N.; Scarlato, G.; Attardi, G. Platelet-mediated transformation of mtDNA-less human cells: Analysis of phenotypic variability among clones from normal individuals—And complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am. J. Hum. Genet. 1994, 54, 966–974. [Google Scholar] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Rieder, M.J.; Taylor, S.L.; Tobe, V.O.; Nickerson, D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: Analysis of the human mitochondrial genome. Nucleic Acids Res. 1998, 26, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Shi, H.; Li, X.; Sun, D.; Li, F.; Li, B.; Ding, Y.; Ma, Y.; Liu, Y.; Zhang, Y.; et al. Exercise intolerance and developmental delay associated with a novel mitochondrial ND5 mutation. Sci. Rep. 2015, 5, 10480. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Ferrin, G.; Perez-Martos, A.; Fernandez-Silva, P.; Zeviani, M.; Enriquez, J.A. Isolation of mitochondria for biogenetical studies: An update. Mitochondrion 2009, 10, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.P.; Schagger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Sharma, L.K.; Fang, H.; Liu, J.; Vartak, R.; Deng, J.; Bai, Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum. Mol. Genet. 2011, 20, 4605–4616. [Google Scholar] [CrossRef] [PubMed]
- Clements, G.B.; Fenyo, E.M.; Klein, G. In vitro derived mouse A9 cell clones differing in malignancy: Analysis by somatic cell hybridization with YACIR lymphoma cell clones. Proc. Natl. Acad. Sci. USA 1976, 73, 2004–2007. [Google Scholar] [CrossRef] [PubMed]
- Rahbari, R.; Sheahan, T.; Modes, V.; Collier, P.; Macfarlane, C.; Badge, R.M. A novel L1 retrotransposon marker for HeLa cell line identification. BioTechniques 2009, 46, 277–284. [Google Scholar] [PubMed]
- Luu, H.H.; Kang, Q.; Park, J.K.; Si, W.; Luo, Q.; Jiang, W.; Yin, H.; Montag, A.G.; Simon, M.A.; Peabody, T.D.; et al. An orthotopic model of human osteosarcoma growth and spontaneous pulmonary metastasis. Clin. Exp. Metastasis 2005, 22, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Farina, A.R.; Tacconelli, A.; Cappabianca, L.; Gulino, A.; Mackay, A.R. Inhibition of human MDA-MB-231 breast cancer cell invasion by matrix metalloproteinase 3 involves degradation of plasminogen. Eur. J. Biochem. FEBS 2002, 269, 4476–4483. [Google Scholar] [CrossRef]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, D.; Li, B.; Qiu, R.; Fang, H.; Lyu, J. Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization. Int. J. Mol. Sci. 2016, 17, 926. https://doi.org/10.3390/ijms17060926
Sun D, Li B, Qiu R, Fang H, Lyu J. Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization. International Journal of Molecular Sciences. 2016; 17(6):926. https://doi.org/10.3390/ijms17060926
Chicago/Turabian StyleSun, Dayan, Bin Li, Ruyi Qiu, Hezhi Fang, and Jianxin Lyu. 2016. "Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization" International Journal of Molecular Sciences 17, no. 6: 926. https://doi.org/10.3390/ijms17060926
APA StyleSun, D., Li, B., Qiu, R., Fang, H., & Lyu, J. (2016). Cell Type-Specific Modulation of Respiratory Chain Supercomplex Organization. International Journal of Molecular Sciences, 17(6), 926. https://doi.org/10.3390/ijms17060926