
Role of mTOR Inhibitors in Kidney Disease



Abstract
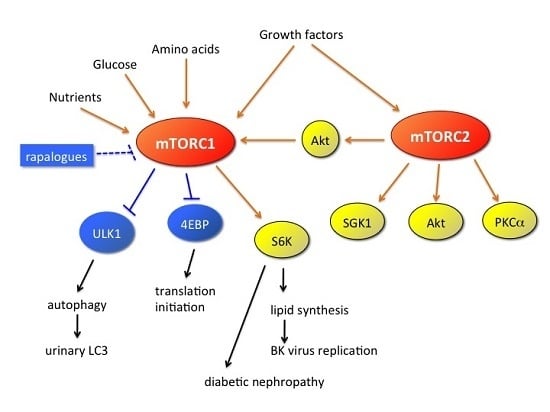
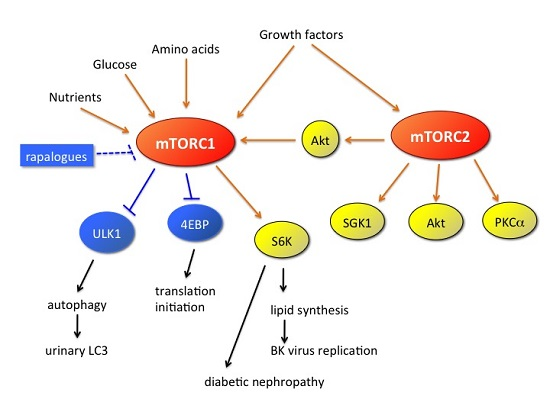
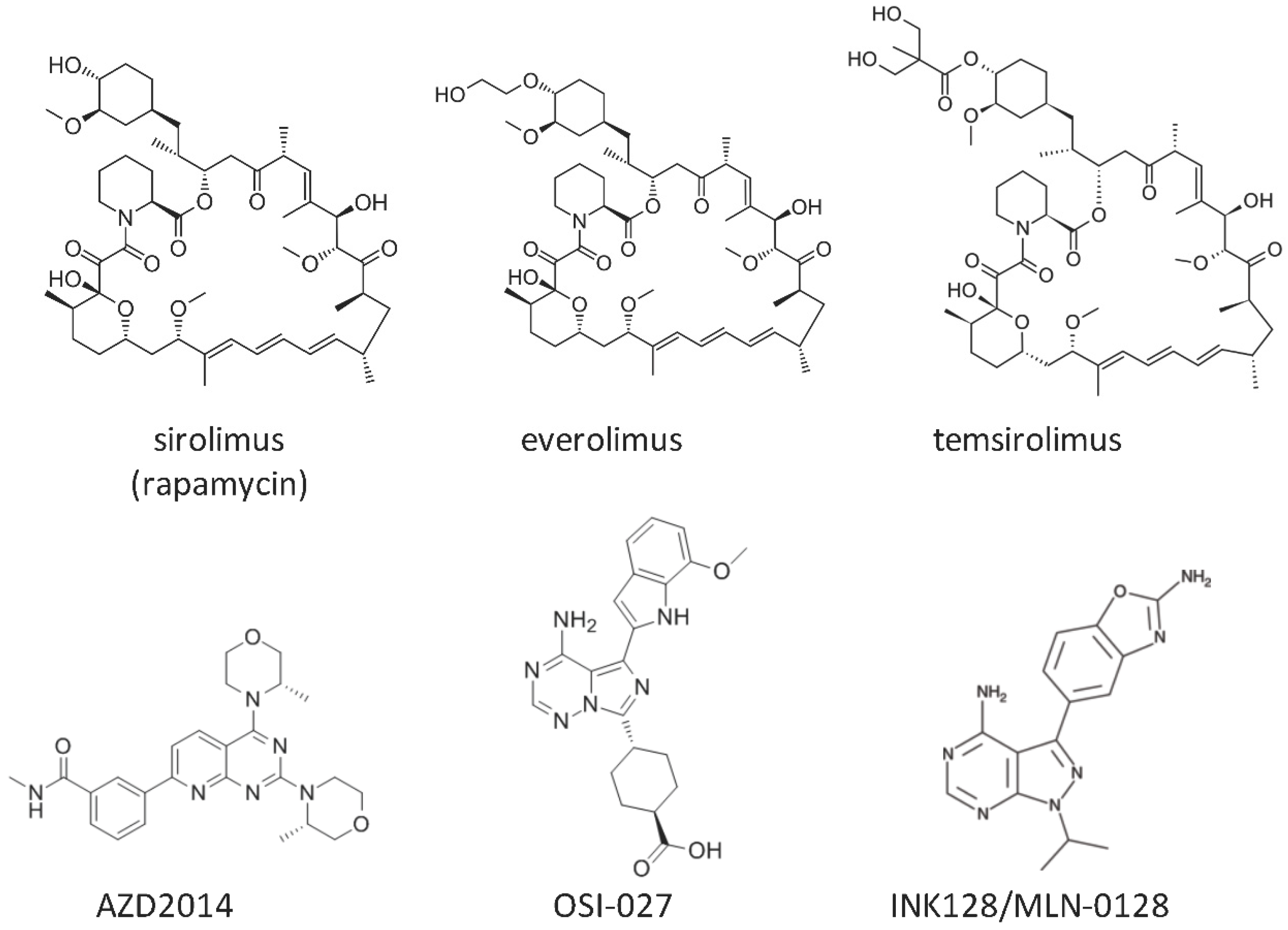
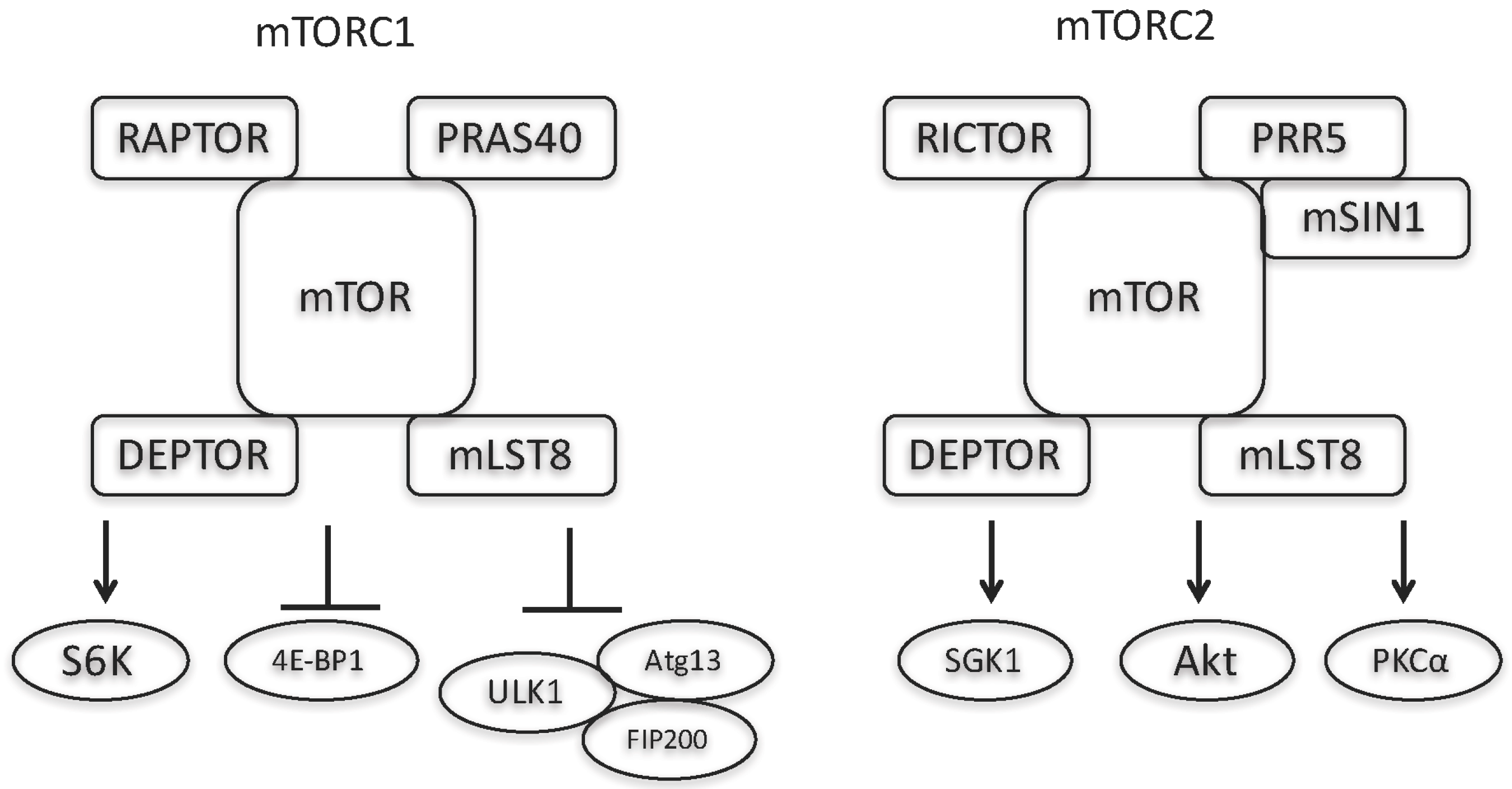
:
{kind=link}
{kind=link}
{kind=link}
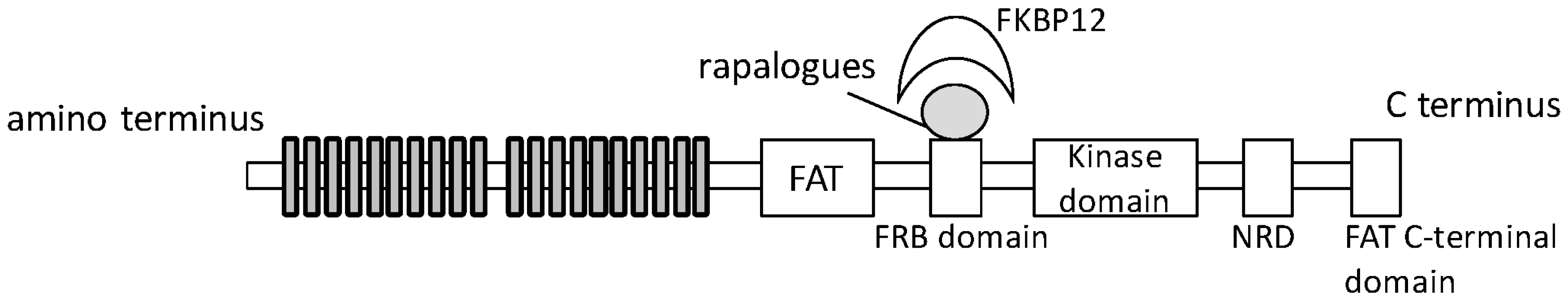{kind=link}
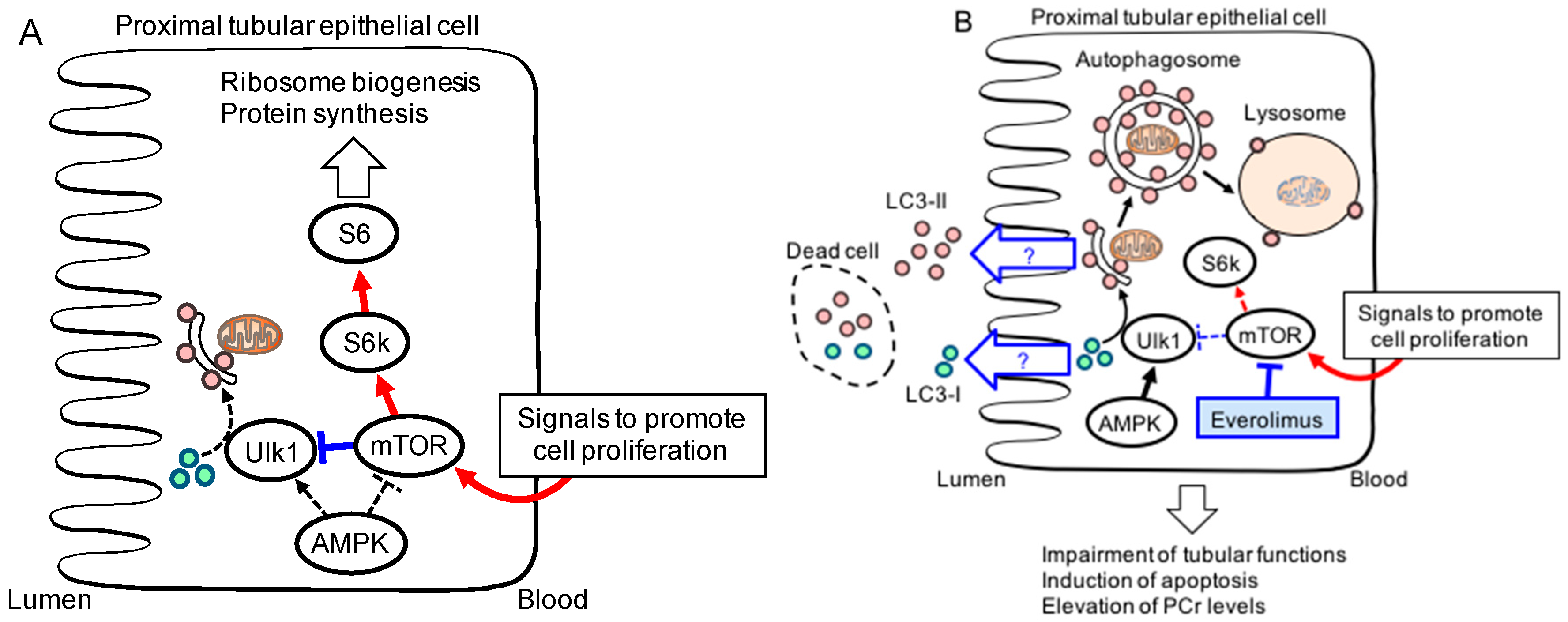{kind=link}
1. Introduction
2. Effect of mTOR Inhibitors on Infection after Kidney Transplantation
3. Urinary Microtubule-Associated Protein 1 Light Chain (LC) 3: A Potential Biomarker for mTOR Inhibition in the Kidneys
4. Application of mTOR Inhibitors in Kidney Transplantation
5. mTOR Inhibitors as Anticancer Drugs
6. Effect of mTOR Inhibitors on Non-Clear Cell Renal Cell Carcinoma
7. Effect of mTOR Inhibitors on Diabetic Nephropathy
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Clippinger, A.J.; Maguire, T.G.; Alwine, J.C. The changing role of mTOR kinase in the maintenance of protein synthesis during human cytomegalovirus infection. J. Virol. 2011, 85, 3930–3939. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Sarbassov dos, D.; Samudio, I.J.; Yee, K.W.; Munsell, M.F.; Ellen Jackson, C.; Giles, F.J.; Sabatini, D.M.; Andreeff, M.; Konopleva, M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007, 109, 3509–3512. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Waldner, M.; Fantus, D.; Solari, M.; Thomson, A.W. New perspectives on mTOR inhibitors (Rapamycin, Rapalogs and TORKinibs) in transplantation. Br. J. Clin. Pharmacol. 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I. Temsirolimus, an inhibitor of mammalian target of rapamycin. Clin Cancer Res 2008, 14, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Moes, D.J.; Guchelaar, H.J.; de Fijter, J.W. Sirolimus and everolimus in kidney transplantation. Drug Discov. Today 2015, 20, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013, 340, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bohl, D.L.; Brennan, D.C. BK virus nephropathy and kidney transplantation. Clin. J. Am. Soc. Nephrol. 2007, 2 (Suppl. 1), S36–S46. [Google Scholar] [CrossRef] [PubMed]
- Jamboti, J.S. BK virus nephropathy in renal transplant recipients. Nephrology (Carlton) 2016, in press. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Knowles, W.; Dickenmann, M.; Passweg, J.; Klimkait, T.; Mihatsch, M.J.; Steiger, J. Prospective study of polyomavirus type BK replication and nephropathy in renal-transplant recipients. N. Engl. J. Med. 2002, 347, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Moscarelli, L.; Caroti, L.; Antognoli, G.; Zanazzi, M.; di Maria, L.; Carta, P.; Minetti, E. Everolimus leads to a lower risk of BKV viremia than mycophenolic acid in de novo renal transplantation patients: A single-center experience. Clin. Transplant. 2013, 27, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wu, H.H.; Weng, C.H.; Chen, Y.C.; Hung, C.C.; Yang, C.W.; Wang, R.Y.; Sakamoto, N.; Tian, Y.C. Cyclophilin A and nuclear factor of activated T cells are essential in cyclosporine-mediated suppression of polyomavirus BK replication. Am. J. Transplant. 2012, 12, 2348–2362. [Google Scholar] [CrossRef] [PubMed]
- Dharnidharka, V.R.; Cherikh, W.S.; Abbott, K.C. An OPTN analysis of national registry data on treatment of BK virus allograft nephropathy in the United States. Transplantation 2009, 87, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Yakhontova, K.; Lu, M.; Manzetti, J. BK Polyomavirus Replication in Renal Tubular Epithelial Cells Is Inhibited by Sirolimus, but activated by tacrolimus through a pathway involving FKBP-12. Am. J. Transplant. 2016, 16, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Liacini, A.; Seamone, M.E.; Muruve, D.A.; Tibbles, L.A. Anti-BK virus mechanisms of sirolimus and leflunomide alone and in combination: Toward a new therapy for BK virus infection. Transplantation 2010, 90, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, D.W.; Chandran, S.; Vincenti, F. Everolimus Conversion to Treat BK Virus Infection in Kidney Transplant Recipients. In Proceedings of the Meeting: 2015 American Transplant Congress, Location of the Conference, Philadelphia, PA, USA, 2–6 May 2015.
- Nashan, B.; Gaston, R.; Emery, V.; Saemann, M.D.; Mueller, N.J.; Couzi, L.; Dantal, J.; Shihab, F.; Mulgaonkar, S.; Seun Kim, Y.; et al. Review of cytomegalovirus infection findings with mammalian target of rapamycin inhibitor-based immunosuppressive therapy in de novo renal transplant recipients. Transplantation 2012, 93, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Ekberg, H.; Tedesco-Silva, H.; Demirbas, A.; Vitko, S.; Nashan, B.; Gurkan, A.; Margreiter, R.; Hugo, C.; Grinyo, J.M.; Frei, U.; et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N. Engl. J. Med. 2007, 357, 2562–2575. [Google Scholar] [CrossRef] [PubMed]
- San Juan, R.; Aguado, J.M.; Lumbreras, C.; Fortun, J.; Munoz, P.; Gavalda, J.; Lopez-Medrano, F.; Montejo, M.; Bou, G.; Blanes, M.; et al. Impact of current transplantation management on the development of cytomegalovirus disease after renal transplantation. Clin. Infect. Dis. 2008, 47, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Taal, M.W.; Luyckx, V.A.; Brenner, B.M. Adaptation to Nephron Loss. In Brenner and Rector’s the Kidney, 7th ed.; Saunders: Philadelphia, PA, USA, 2004. [Google Scholar]
- Pontrelli, P.; Rossini, M.; Infante, B.; Stallone, G.; Schena, A.; Loverre, A.; Ursi, M.; Verrienti, R.; Maiorano, A.; Zaza, G.; et al. Rapamycin inhibits PAI-1 expression and reduces interstitial fibrosis and glomerulosclerosis in chronic allograft nephropathy. Transplantation 2008, 85, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Stallone, G.; Infante, B.; Schena, A.; Battaglia, M.; Ditonno, P.; Loverre, A.; Gesualdo, L.; Schena, F.P.; Grandaliano, G. Rapamycin for treatment of chronic allograft nephropathy in renal transplant patients. J. Am. Soc. Nephrol. 2005, 16, 3755–3762. [Google Scholar] [CrossRef] [PubMed]
- Lloberas, N.; Cruzado, J.M.; Franquesa, M.; Herrero-Fresneda, I.; Torras, J.; Alperovich, G.; Rama, I.; Vidal, A.; Grinyo, J.M. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J. Am. Soc. Nephrol. 2006, 17, 1395–1404. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Matsubara, T.; Mima, A.; Sumi, E.; Kanamori, H.; Iehara, N.; Fukatsu, A.; Yanagita, M.; Nakano, T.; Ishimoto, Y.; et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int. 2005, 68, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef] [PubMed]
- Huber, T.B.; Edelstein, C.L.; Hartleben, B.; Inoki, K.; Jiang, M.; Jiang, M.; Koya, D.; Kume, S.; Lieberthal, W.; Pallet, N.; Pallet, N.; et al. Emerging role of autophagy in kidney function, diseases and aging Autophagy in acute kidney injury Role of mTOR in podocyte function and diabetic nephropathy in humans and mice mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. Autophagy 2012, 8, 1009–1031. [Google Scholar] [PubMed]
- Nakagawa, S.; Masuda, S.; Nishihara, K.; Inui, K. mTOR inhibitor everolimus ameliorates progressive tubular dysfunction in chronic renal failure rats. Biochem. Pharmacol. 2010, 79, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Kaushal, G.P.; Shah, S.V. Autophagy in acute kidney injury. Kidney Int. 2016, 89, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Nishihara, K.; Inui, K.; Masuda, S. Involvement of autophagy in the pharmacological effects of the mTOR inhibitor everolimus in acute kidney injury. Eur. J. Pharmacol. 2012, 696, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Rostaing, L.; Christiaans, M.H.; Kovarik, J.M.; Pascual, J. The pharmacokinetics of everolimus in de novo kidney transplant patients receiving tacrolimus: An analysis from the randomized ASSET study. Ann. Transplant. 2014, 19, 337–345. [Google Scholar] [PubMed]
- Naesens, M.; Kuypers, D.R.; Sarwal, M. Calcineurin inhibitor nephrotoxicity. Clin. J. Am. Soc. Nephrol 2009, 4, 481–508. [Google Scholar] [CrossRef] [PubMed]
- Pape, L.; Ahlenstiel, T. mTOR inhibitors in pediatric kidney transplantation. Pediatr. Nephrol. 2014, 29, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Escuredo, A.; Diekmann, F.; Revuelta, I.; Esforzado, N.; Ricart, M.J.; Cofan, F.; Torregrosa, J.V.; Peri, L.; Ruiz, A.; Campistol, J.M.; et al. An mTOR-inhibitor-based protocol and calcineurin inhibitor (CNI)-free treatment in kidney transplant recipients from donors after cardiac death: Good renal function, but high incidence of conversion to CNI. Transpl. Int. 2016, 29, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.J.; Villa, M.; Laskey, R.; Benavides, C.; Schoenberg, L.; Welsh, M.; Kerman, R.H.; Podder, H.; van Buren, C.T.; Katz, S.M.; et al. Risk factors for impaired wound healing in sirolimus-treated renal transplant recipients. Clin. Transplant. 2007, 21, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Santoni, M.; Medri, M.; Scarpi, E.; Burattini, L.; Lolli, C.; Rossi, L.; Savini, A.; Berardi, R.; Stanganelli, I.; et al. Correlation of stomatitis and cutaneous toxicity with clinical outcome in patients with metastatic renal-cell carcinoma treated with everolimus. Clin. Genitourin Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Grenda, R.; Watson, A.; Trompeter, R.; Tonshoff, B.; Jaray, J.; Fitzpatrick, M.; Murer, L.; Vondrak, K.; Maxwell, H.; van Damme-Lombaerts, R.; et al. A randomized trial to assess the impact of early steroid withdrawal on growth in pediatric renal transplantation: The TWIST study. Am. J. Transplant. 2010, 10, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Chan, H.Y.; Grossman, A.B.; Bukowski, R.M. Everolimus in the treatment of renal cell carcinoma and neuroendocrine tumors. Adv. Ther. 2010, 27, 495–511. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.H.; Chadalavada, R.S.; Chaganti, R.S.; Motzer, R.J. Targeting von Hippel-Lindau pathway in renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7215–7220. [Google Scholar] [CrossRef] [PubMed]
- Masoud, G.N.; Li, W. HIF-1alpha pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Schmidinger, M. Improving outcomes in metastatic clear cell renal cell carcinoma by sequencing therapy. Am. Soc. Clin. Oncol. Educ. Book 2014, e228–e238. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Jonasch, E.; Agarwal, N.; Beard, C.; Bhayani, S.; Bolger, G.B.; Chang, S.S.; Choueiri, T.K.; Costello, B.A.; Derweesh, I.H.; et al. Kidney cancer, version 3.2015. J. Natl. Compr. Canc. Netw. 2015, 13, 151–159. [Google Scholar] [PubMed]
- Patel, S.B.; Stenehjem, D.D.; Gill, D.M.; Tantravahi, S.K.; Agarwal, A.M.; Hsu, J.; Vuong, W.; Pal, S.K.; Agarwal, N. Everolimus versus temsirolimus in metastatic renal cell carcinoma after progression with previous systemic therapies. Clin. Genitourin Cancer 2016, 14, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Iacovelli, R.; Santoni, M.; Verzoni, E.; Grassi, P.; Testa, I.; de Braud, F.; Cascinu, S.; Procopio, G. Everolimus and temsirolimus are not the same second-line in metastatic renal cell carcinoma. A systematic review and meta-analysis of literature data. Clin. Genitourin Cancer 2015, 13, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Eisen, T.; Broderick, S.; Stadler, W.M.; Jones, R.J.; Garcia, J.A.; Vaishampayan, U.N.; Picus, J.; Hawkins, R.E.; et al. Everolimus versus sunitinib for patients with metastatic non-clear cell renal cell carcinoma (ASPEN): A multicentre, open-label, randomised phase 2 trial. Lancet Oncol. 2016, 17, 378–388. [Google Scholar] [CrossRef]
- Tannir, N.M.; Jonasch, E.; Albiges, L.; Altinmakas, E.; Ng, C.S.; Matin, S.F.; Wang, X.; Qiao, W.; Dubauskas Lim, Z.; Tamboli, P.; et al. Everolimus versus sunitinib prospective evaluation in metastatic non-clear cell renal cell carcinoma (ESPN): A randomized multicenter phase 2 trial. Eur. Urol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.Y.; Wu, S. Update on the treatment of metastatic clear cell and non-clear cell renal cell carcinoma. Biomark. Res. 2015, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.; Lim, H.Y.; Ahn, J.H.; Lee, J.L.; Rha, S.Y.; Kim, Y.J.; Kim, T.M.; Lee, S.H. Phase II trial of everolimus for the treatment of nonclear-cell renal cell carcinoma. Ann. Oncol. 2013, 24, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Barrios, C.H.; Kim, T.M.; Falcon, S.; Cosgriff, T.; Harker, W.G.; Srimuninnimit, V.; Pittman, K.; Sabbatini, R.; Rha, S.Y.; et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2014, 32, 2765–2772. [Google Scholar] [CrossRef] [PubMed]
- Verges, B.; Cariou, B. mTOR inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Schena, F.P.; Gesualdo, L. Pathogenetic mechanisms of diabetic nephropathy. J. Am. Soc. Nephrol. 2005, 16 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef] [PubMed]
- Koya, D.; Jirousek, M.R.; Lin, Y.W.; Ishii, H.; Kuboki, K.; King, G.L. Characterization of protein kinase C beta isoform activation on the gene expression of transforming growth factor-beta, extracellular matrix components, and prostanoids in the glomeruli of diabetic rats. J. Clin. Investig. 1997, 100, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Bo, H.; Villani, V.; Spencer, P.J.; Fu, P. The Inhibitory Effect of Rapamycin on Toll Like Receptor 4 and Interleukin 17 in the Early Stage of Rat Diabetic Nephropathy. Kidney Blood Press Res. 2016, 41, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Velagapudi, C.; Bhandari, B.S.; Abboud-Werner, S.; Simone, S.; Abboud, H.E.; Habib, S.L. The tuberin/mTOR pathway promotes apoptosis of tubular epithelial cells in diabetes. J. Am. Soc. Nephrol. 2011, 22, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.; Freilinger, A.; Hengstschlager, M. Akt regulates nuclear/cytoplasmic localization of tuberin. Oncogene 2007, 26, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.A.; Harwood, S.; Varagunam, M.; Raftery, M.J.; Yaqoob, M.M. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J. 2003, 17, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Inoki, K.; Masutani, K.; Wakabayashi, Y.; Komai, K.; Nakagawa, R.; Guan, K.L.; Yoshimura, A. The mTOR pathway is highly activated in diabetic nephropathy and rapamycin has a strong therapeutic potential. Biochem. Biophys. Res. Commun. 2009, 384, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Godel, M.; Hartleben, B.; Herbach, N.; Liu, S.; Zschiedrich, S.; Lu, S.; Debreczeni-Mor, A.; Lindenmeyer, M.T.; Rastaldi, M.P.; Hartleben, G.; et al. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J. Clin. Investig. 2011, 121, 2197–2209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Mori, H.; Wang, J.; Suzuki, T.; Hong, S.; Yoshida, S.; Blattner, S.M.; Ikenoue, T.; Ruegg, M.A.; Hall, M.N.; et al. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J. Clin. Investig. 2011, 121, 2181–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kajiwara, M.; Masuda, S. Role of mTOR Inhibitors in Kidney Disease. Int. J. Mol. Sci. 2016, 17, 975. https://doi.org/10.3390/ijms17060975
Kajiwara M, Masuda S. Role of mTOR Inhibitors in Kidney Disease. International Journal of Molecular Sciences. 2016; 17(6):975. https://doi.org/10.3390/ijms17060975
Chicago/Turabian StyleKajiwara, Moto, and Satohiro Masuda. 2016. "Role of mTOR Inhibitors in Kidney Disease" International Journal of Molecular Sciences 17, no. 6: 975. https://doi.org/10.3390/ijms17060975
APA StyleKajiwara, M., & Masuda, S. (2016). Role of mTOR Inhibitors in Kidney Disease. International Journal of Molecular Sciences, 17(6), 975. https://doi.org/10.3390/ijms17060975