
The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy


Abstract
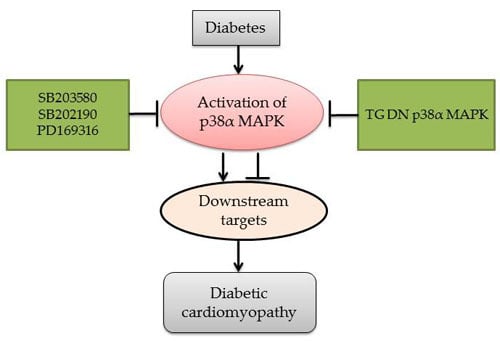
:
1. Introduction
2. Structure and Molecular Biology of p38 Mitogen-Activated Protein Kinase (MAPK)
3. Effect of p38 MAPK Activation on Hearts of Diabetic Individuals
3.1. Inflammatory and Oxidative Stress Pathways
3.2. The Apoptotic Pathway
3.3. Pathological Hypertrophy
3.4. Energy Metabolism Pathway
4. Protective Role of p38β MAPK in Diabetes
5. The Inhibition of p38 MAPK Is Beneficial for Diabetic Complications
5.1. Specific Inhibition of p38 MAPK with Inhibitors
5.2. Suppression of p38α MAPK in Dominant-Negative Mutant of Transgenic Model
6. Suppression of the Downstream of p38 MAPK
7. MicroRNAs Associated with p38 MAPK Activity in Diabetes
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ANP | Atrium natriuretic peptide |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| BNP | Brain natriuretic peptide |
| CHOP | C/EBP homologous protein |
| DCM | Diabetic cardiomyopathy |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| FAK | Focal adhesion kinase |
| FFA | Free fatty acid |
| GLUT4 | Glucose transporter type 4 |
| HG | High glucose |
| IL-6 | Interleukin-6 |
| IRS1 | Insulin receptor substrates 1 |
| IRS2 | Insulin receptor substrates 2 |
| JNK | C-Jun N-terminal protein kinase |
| I.P | Intraperitoneal injection |
| MEF2 | Myocyte enhancer factor 2 |
| MAPK | Mitogen-activated protein kinase |
| MK2 | MAPKAPK-2 |
| MK3 | MAPKAPK-3 |
| NF-κB | Nuclear factor-κB |
| P38 MAPK | P38 MAP kinase |
| PTEN | Phosphatase and tensin homolog |
| ROS | Reactive oxygen species |
| SERCA2a | Sarcoendoplasmic reticulum Ca2+-ATPase 2a |
| SDF-1β | Stromal cell-derived factor-1β |
| SMAD | Similar to mothers against decapentaplegic homolog |
| STAT1 | Signal transducer and activator of transcription 1 |
| STZ | Streptozotocin |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| TNF-α | Tissue necrosis factor α |
| TGF-β | Transforming growth factor-β |
References
- Zhang, Z.; Wang, S.; Zhou, S.; Yan, X.; Wang, Y.; Chen, J.; Mellen, N.; Kong, M.; Gu, J.; Tan, Y.; et al. Sulforaphane prevents the development of cardiomyopathy in type 2 diabetic mice probably by reversing oxidative stress-induced inhibition of LKB1/AMPK pathway. J. Mol. Cell. Cardiol. 2014, 77, 42–52. [Google Scholar]
- Bai, Y.; Cui, W.; Xin, Y.; Miao, X.; Barati, M.T.; Zhang, C.; Chen, Q.; Tan, Y.; Cui, T.; Zheng, Y.; et al. Prevention by sulforaphane of diabetic cardiomyopathy is associated with up-regulation of NRF2 expression and transcription activation. J. Mol. Cell. Cardiol. 2013, 57, 82–95. [Google Scholar]
- Zhang, C.; Huang, Z.; Gu, J.; Yan, X.; Lu, X.; Zhou, S.; Wang, S.; Shao, M.; Zhang, F.; Cheng, P.; et al. Fibroblast growth factor 21 protects the heart from apoptosis in a diabetic mouse model via extracellular signal-regulated kinase 1/2-dependent signalling pathway. Diabetologia 2015, 58, 1937–1948. [Google Scholar]
- Oh, C.C.; Nguy, M.Q.; Schwenke, D.C.; Migrino, R.Q.; Thornburg, K.; Reaven, P. P38α mitogen-activated kinase mediates cardiomyocyte apoptosis induced by palmitate. Biochem. Biophys. Res. Commun. 2014, 450, 628–633. [Google Scholar]
- Zhang, C.; Zhang, L.; Chen, S.; Feng, B.; Lu, X.; Bai, Y.; Liang, G.; Tan, Y.; Shao, M.; Skibba, M.; et al. The prevention of diabetic cardiomyopathy by non-mitogenic acidic fibroblast growth factor is probably mediated by the suppression of oxidative stress and damage. PLoS ONE 2013, 8, e82287. [Google Scholar]
- Zhao, Y.; Tang, Z.; Zhu, X.; Wang, X.; Wang, C.; Zhang, W.; Xia, N.; Wang, S.; Huang, J.; Cui, S. TAB3 involves in hepatic insulin resistance through activation of MAPK pathway. Gen. Comp. Endocrinol. 2015, 224, 228–234. [Google Scholar]
- Dadson, K.; Turdi, S.; Hashemi, S.; Zhao, J.; Polidovitch, N.; Beca, S.; Backx, P.H.; McDermott, J.C.; Sweeney, G. Adiponectin is required for cardiac MEF2 activation during pressure overload induced hypertrophy. J. Mol. Cell. Cardiol. 2015, 86, 102–109. [Google Scholar]
- Gao, Y.; Kang, L.; Li, C.; Wang, X.; Sun, C.; Li, Q.; Liu, R.; Wang, J. Resveratrol ameliorates diabetes-induced cardiac dysfunction through AT1R-ERK/p38 MAPK signaling pathway. Cardiovasc. Toxicol. 2016, 16, 130–137. [Google Scholar]
- Thandavarayan, R.A.; Giridharan, V.V.; Arumugam, S.; Suzuki, K.; Ko, K.M.; Krishnamurthy, P.; Watanabe, K.; Konishi, T. Schisandrin b prevents doxorubicin induced cardiac dysfunction by modulation of DNA damage, oxidative stress and inflammation through inhibition of MAPK/p53 signaling. PLoS ONE 2015, 10, e0119214. [Google Scholar]
- Kim, M.; Oh, J.K.; Sakata, S.; Liang, I.; Park, W.; Hajjar, R.J.; Lebeche, D. Role of resistin in cardiac contractility and hypertrophy. J. Mol. Cell. Cardiol. 2008, 45, 270–280. [Google Scholar]
- Pan, Y.; Wang, Y.; Zhao, Y.; Peng, K.; Li, W.; Wang, Y.; Zhang, J.; Zhou, S.; Liu, Q.; Li, X.; et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes 2014, 63, 3497–3511. [Google Scholar]
- Yan, Z.; Ni, Y.; Wang, P.; Chen, J.; He, H.; Sun, J.; Cao, T.; Chen, J.; Zhao, Z.; Luo, Z.; et al. Peroxisome proliferator-activated receptor delta protects against obesity-related glomerulopathy through the p38 MAPK pathway. Obesity 2013, 21, 538–545. [Google Scholar]
- Zhu, S.; Yang, Y.; Hu, J.; Qian, L.; Jiang, Y.; Li, X.; Yang, Q.; Bai, H.; Chen, Q. Wld(s) ameliorates renal injury in a type 1 diabetic mouse model. Am. J. Physiol. Ren. Physiol. 2014, 306, F1348–F1356. [Google Scholar]
- Cao, L.; Qin, X.; Peterson, M.R.; Haller, S.E.; Wilson, K.A.; Hu, N.; Lin, X.; Nair, S.; Ren, J.; He, G. CARD9 knockout ameliorates myocardial dysfunction associated with high fat diet-induced obesity. J. Mol. Cell. Cardiol. 2016, 92, 185–195. [Google Scholar]
- Westermann, D.; Rutschow, S.; van Linthout, S.; Linderer, A.; Bucker-Gartner, C.; Sobirey, M.; Riad, A.; Pauschinger, M.; Schultheiss, H.P.; Tschope, C. Inhibition of p38 mitogen-activated protein kinase attenuates left ventricular dysfunction by mediating pro-inflammatory cardiac cytokine levels in a mouse model of diabetes mellitus. Diabetologia 2006, 49, 2507–2513. [Google Scholar]
- Kassan, M.; Choi, S.K.; Galan, M.; Lee, Y.H.; Trebak, M.; Matrougui, K. Enhanced p22phox expression impairs vascular function through p38 and ERK1/2 map kinase-dependent mechanisms in type 2 diabetic mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H972–H980. [Google Scholar]
- Yokota, T.; Wang, Y. P38 map kinases in the heart. Gene 2016, 575, 369–376. [Google Scholar]
- Lemke, L.E.; Bloem, L.J.; Fouts, R.; Esterman, M.; Sandusky, G.; Vlahos, C.J. Decreased p38 MAPK activity in end-stage failing human myocardium: P38 MAPKα is the predominant isoform expressed in human heart. J. Mol. Cell. Cardiol. 2001, 33, 1527–1540. [Google Scholar]
- Koivisto, E.; Kaikkonen, L.; Tokola, H.; Pikkarainen, S.; Aro, J.; Pennanen, H.; Karvonen, T.; Rysa, J.; Kerkela, R.; Ruskoaho, H. Distinct regulation of b-type natriuretic peptide transcription by p38 MAPK isoforms. Mol. Cell. Endocrinol. 2011, 338, 18–27. [Google Scholar]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar]
- Allen, M.; Svensson, L.; Roach, M.; Hambor, J.; McNeish, J.; Gabel, C.A. Deficiency of the stress kinase p38α results in embryonic lethality: Characterization of the kinase dependence of stress responses of enzyme-deficient embryonic stem cells. J. Exp. Med. 2000, 191, 859–870. [Google Scholar]
- Taniike, M.; Yamaguchi, O.; Tsujimoto, I.; Hikoso, S.; Takeda, T.; Nakai, A.; Omiya, S.; Mizote, I.; Nakano, Y.; Higuchi, Y.; et al. Apoptosis signal-regulating kinase 1/p38 signaling pathway negatively regulates physiological hypertrophy. Circulation 2008, 117, 545–552. [Google Scholar]
- Adams, R.H.; Porras, A.; Alonso, G.; Jones, M.; Vintersten, K.; Panelli, S.; Valladares, A.; Perez, L.; Klein, R.; Nebreda, A.R. Essential role of p38α MAP kinase in placental but not embryonic cardiovascular development. Mol. Cell 2000, 6, 109–116. [Google Scholar]
- O’Keefe, S.J.; Mudgett, J.S.; Cupo, S.; Parsons, J.N.; Chartrain, N.A.; Fitzgerald, C.; Chen, S.L.; Lowitz, K.; Rasa, C.; Visco, D.; et al. Chemical genetics define the roles of p38α and p38β in acute and chronic inflammation. J. Biol. Chem. 2007, 282, 34663–34671. [Google Scholar]
- Xie, J.; He, G.; Chen, Q.; Sun, J.; Dai, Q.; Lu, J.; Li, G.; Wu, H.; Li, R.; Chen, J.; et al. Syndecan 4 signaling is required for exercise-induced cardiac hypertrophy. Mol. Med. 2016. [Google Scholar] [CrossRef]
- Watanabe, K.; Ma, M.; Hirabayashi, K.; Gurusamy, N.; Veeraveedu, P.T.; Prakash, P.; Zhang, S.; Muslin, A.J.; Kodama, M.; Aizawa, Y. Swimming stress in DN 14-3-3 mice triggers maladaptive cardiac remodeling: Role of p38 MAPK. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1269–H1277. [Google Scholar]
- Zabalgoitia, M.; Colston, J.T.; Reddy, S.V.; Holt, J.W.; Regan, R.F.; Stec, D.E.; Rimoldi, J.M.; Valente, A.J.; Chandrasekar, B. Carbon monoxide donors or heme oxygenase-1 (HO-1) overexpression blocks interleukin-18-mediated NF-κB-PTEN-dependent human cardiac endothelial cell death. Free Radic. Biol. Med. 2008, 44, 284–298. [Google Scholar]
- Liu, H.; Pedram, A.; Kim, J.K. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38α-mediated activation of p53 and by down-regulating p53 inhibition on p38β. Cardiovasc. Res. 2011, 89, 119–128. [Google Scholar]
- Thandavarayan, R.A.; Watanabe, K.; Ma, M.; Gurusamy, N.; Veeraveedu, P.T.; Konishi, T.; Zhang, S.; Muslin, A.J.; Kodama, M.; Aizawa, Y. Dominant-negative p38α mitogen-activated protein kinase prevents cardiac apoptosis and remodeling after streptozotocin-induced diabetes mellitus. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H911–H919. [Google Scholar]
- Ruiz, M.; Coderre, L.; Lachance, D.; Houde, V.; Martel, C.; Legault, J.T.; Gillis, M.A.; Bouchard, B.; Daneault, C.; Carpentier, A.C.; et al. MK2 deletion in mice prevents diabetes-induced perturbations in lipid metabolism and cardiac dysfunction. Diabetes. 2016, 65, 381–392. [Google Scholar]
- Liu, Z.; Cao, W. P38 mitogen-activated protein kinase: A critical node linking insulin resistance and cardiovascular diseases in type 2 diabetes mellitus. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 38–46. [Google Scholar]
- Liang, Z.; Leo, S.; Wen, H.; Ouyang, M.; Jiang, W.; Yang, K. Triptolide improves systolic function and myocardial energy metabolism of diabetic cardiomyopathy in streptozotocin-induced diabetic rats. BMC Cardiovasc. Disord. 2015, 15, 42. [Google Scholar] [CrossRef]
- Zhenzhong, Z.; Yafa, Y.; Jin, L. Fibrinogen-like protein 2 gene silencing inhibits cardiomyocytes apoptosis, improves heart function of streptozotocin-induced diabetes rats and the molecular mechanism involved. Biosci Rep. 2015, 35. [Google Scholar] [CrossRef]
- Hao, P.; Yang, J.; Liu, Y.; Zhang, M.; Zhang, K.; Gao, F.; Chen, Y.; Zhang, C.; Zhang, Y. Combination of angiotensin-(1–7) with perindopril is better than single therapy in ameliorating diabetic cardiomyopathy. Sci. Rep. 2015, 5, 8794. [Google Scholar]
- Li, C.J.; Lv, L.; Li, H.; Yu, D.M. Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha-lipoic acid. Cardiovasc. Diabetol. 2012, 11, 73. [Google Scholar] [CrossRef]
- Yu, W.; Zha, W.; Guo, S.; Cheng, H.; Wu, J.; Liu, C. Flos puerariae extract prevents myocardial apoptosis via attenuation oxidative stress in streptozotocin-induced diabetic mice. PLoS ONE 2014, 9, e98044. [Google Scholar]
- Gilardini Montani, M.S.; Granato, M.; Cuomo, L.; Sandro, V.; di Renzo, L.; D’Orazi, G.; Faggioni, A.; Cirone, M. High glucose and hyperglycemic sera from type 2 diabetic patients impair dc differentiation by inducing ros and activating wnt/beta-catenin and p38 MAPK. Biochim. Biophys. Acta 2016, 1862, 805–813. [Google Scholar]
- Ren, Y.; Shi, Y.; Wang, Y.; Li, Y.; Wu, S.; Li, H.; Zhang, Y.; Duan, H. P38 MAPK pathway is involved in high glucose-induced thioredoxin interacting protein induction in mouse mesangial cells. FEBS Lett. 2010, 584, 3480–3485. [Google Scholar]
- Ki, Y.W.; Park, J.H.; Lee, J.E.; Shin, I.C.; Koh, H.C. JNK and p38 MAPK regulate oxidative stress and the inflammatory response in chlorpyrifos-induced apoptosis. Toxicol. Lett. 2013, 218, 235–245. [Google Scholar]
- Boudina, S.; Abel, E.D. Diabetic cardiomyopathy revisited. Circulation 2007, 115, 3213–3223. [Google Scholar]
- Zhao, Y.; Tan, Y.; Xi, S.; Li, Y.; Li, C.; Cui, J.; Yan, X.; Li, X.; Wang, G.; Li, W.; et al. A novel mechanism by which SDF-1β protects cardiac cells from palmitate-induced endoplasmic reticulum stress and apoptosis via CXCR7 and AMPK/p38 MAPK-mediated interleukin-6 generation. Diabetes 2013, 62, 2545–2558. [Google Scholar]
- Yan, J.; Young, M.E.; Cui, L.; Lopaschuk, G.D.; Liao, R.; Tian, R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 2009, 119, 2818–2828. [Google Scholar]
- Maillet, M.; van Berlo, J.H.; Molkentin, J.D. Molecular basis of physiological heart growth: Fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013, 14, 38–48. [Google Scholar]
- Hsieh, Y.L.; Tsai, Y.L.; Shibu, M.A.; Su, C.C.; Chung, L.C.; Pai, P.; Kuo, C.H.; Yeh, Y.L.; Viswanadha, V.P.; Huang, C.Y. ZAK induces cardiomyocyte hypertrophy and brain natriuretic peptide expression via p38/JNK signaling and GATA4/c-Jun transcriptional factor activation. Mol. Cell. Biochem. 2015, 405, 1–9. [Google Scholar]
- Hu, W.S.; Ho, T.J.; Pai, P.; Chung, L.C.; Kuo, C.H.; Chang, S.H.; Tsai, F.J.; Tsai, C.H.; Jie, Y.C.; Liou, Y.M.; et al. Gelsolin (GSN) induces cardiomyocyte hypertrophy and BNP expression via p38 signaling and GATA-4 transcriptional factor activation. Mol. Cell. Biochem. 2014, 390, 263–270. [Google Scholar]
- Feng, B.; Chen, S.; George, B.; Feng, Q.; Chakrabarti, S. Mir133a regulates cardiomyocyte hypertrophy in diabetes. Diabetes Metab. Res. Rev. 2010, 26, 40–49. [Google Scholar]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar]
- Harmancey, R.; Wilson, C.R.; Taegtmeyer, H. Adaptation and maladaptation of the heart in obesity. Hypertension 2008, 52, 181–187. [Google Scholar]
- Zhang, K.; Li, L.; Qi, Y.; Zhu, X.; Gan, B.; DePinho, R.A.; Averitt, T.; Guo, S. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012, 153, 631–646. [Google Scholar]
- Qi, Y.; Xu, Z.; Zhu, Q.; Thomas, C.; Kumar, R.; Feng, H.; Dostal, D.E.; White, M.F.; Baker, K.M.; Guo, S. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes 2013, 62, 3887–3900. [Google Scholar]
- Battiprolu, P.K.; Hojayev, B.; Jiang, N.; Wang, Z.V.; Luo, X.; Iglewski, M.; Shelton, J.M.; Gerard, R.D.; Rothermel, B.A.; Gillette, T.G.; et al. Metabolic stress-induced activation of Foxo1 triggers diabetic cardiomyopathy in mice. J. Clin. Investig. 2012, 122, 1109–1118. [Google Scholar]
- Evans-Anderson, H.J.; Alfieri, C.M.; Yutzey, K.E. Regulation of cardiomyocyte proliferation and myocardial growth during development by Foxo transcription factors. Circ. Res. 2008, 102, 686–694. [Google Scholar]
- Venkatakrishnan, C.D.; Tewari, A.K.; Moldovan, L.; Cardounel, A.J.; Zweier, J.L.; Kuppusamy, P.; Ilangovan, G. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2680–H2691. [Google Scholar]
- Van Linthout, S.; Riad, A.; Dhayat, N.; Spillmann, F.; Du, J.; Dhayat, S.; Westermann, D.; Hilfiker-Kleiner, D.; Noutsias, M.; Laufs, U.; et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia 2007, 50, 1977–1986. [Google Scholar]
- Zuo, L.; Du, Y.; Lu, M.; Gao, J.; Hu, R.; Zhang, S.; Wang, Y.; Zhu, H.; Zhou, Q.; Wei, W.; et al. Atorvastatin inhibits hyperglycemia-induced expression of osteopontin in the diabetic rat kidney via the p38 MAPK pathway. Mol. Biol. Rep. 2014, 41, 2551–2558. [Google Scholar]
- Ahad, A.; Ahsan, H.; Mujeeb, M.; Siddiqui, W.A. Gallic acid ameliorates renal functions by inhibiting the activation of p38 MAPK in experimentally induced type 2 diabetic rats and cultured rat proximal tubular epithelial cells. Chem. Biol. Interact. 2015, 240, 292–303. [Google Scholar]
- Lv, G.F.; Dong, M.L.; Hu, D.H.; Zhang, W.F.; Wang, Y.C.; Tang, C.W.; Zhu, X.X. Insulin-mediated inhibition of p38 mitogen-activated protein kinase protects cardiomyocytes in severe burns. J. Burn. Care Res. 2011, 32, 591–599. [Google Scholar]
- Fang, D.; Guan, H.; Liu, J.; Wei, G.; Ke, W.; Yao, B.; Xiao, H.; Li, Y. Early intensive insulin therapy attenuates the p38 pathway in the renal cortex and indices of nephropathy in diabetic rats. Endocr. J. 2012, 59, 81–90. [Google Scholar]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar]
- Komers, R.; Schutzer, W.; Xue, H.; Oyama, T.T.; Lindsley, J.N.; Anderson, S. Effects of p38 mitogen-activated protein kinase inhibition on blood pressure, renal hemodynamics, and renal vascular reactivity in normal and diabetic rats. Transl. Res. 2007, 150, 343–349. [Google Scholar]
- Higham, A.; Lea, S.; Ray, D.; Singh, D. Corticosteroid effects on copd alveolar macrophages: Dependency on cell culture methodology. J. Immunol. Methods 2014, 405, 144–153. [Google Scholar]
- Devaraj, S.; Venugopal, S.K.; Singh, U.; Jialal, I. Hyperglycemia induces monocytic release of interleukin-6 via induction of protein kinase c-α and -β. Diabetes 2005, 54, 85–91. [Google Scholar]
- Turner, N.A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Modulatory effect of interleukin-1α on expression of structural matrix proteins, mmps and timps in human cardiac myofibroblasts: Role of p38 map kinase. Matrix Biol. 2010, 29, 613–620. [Google Scholar]
- American Diabetes Association. Diagnosis and classification of diabetes mellitus. Diabetes Care 2014, 37, S81–S90. [Google Scholar]
- Cheung, P.C.; Campbell, D.G.; Nebreda, A.R.; Cohen, P. Feedback control of the protein kinase TAK1 by SAPK2A/p38α. Embo J. 2003, 22, 5793–5805. [Google Scholar]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. P38(MAPK): Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat. Med. 2005, 11, 183–190. [Google Scholar]
- Ozcan, L.; Cristina de Souza, J.; Harari, A.A.; Backs, J.; Olson, E.N.; Tabas, I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013, 18, 803–815. [Google Scholar]
- Thandavarayan, R.A.; Giridharan, V.V.; Sari, F.R.; Arumugam, S.; Veeraveedu, P.T.; Pandian, G.N.; Palaniyandi, S.S.; Ma, M.; Suzuki, K.; Gurusamy, N.; et al. Depletion of 14-3-3 protein exacerbates cardiac oxidative stress, inflammation and remodeling process via modulation of MAPK/NF-κB signaling pathways after streptozotocin-induced diabetes mellitus. Cell. Physiol. Biochem. 2011, 28, 911–922. [Google Scholar]
- Ozcan, L.; Xu, X.; Deng, S.X.; Ghorpade, D.S.; Thomas, T.; Cremers, S.; Hubbard, B.; Serrano-Wu, M.H.; Gaestel, M.; Landry, D.W.; et al. Treatment of obese insulin-resistant mice with an allosteric MAPKAPK2/3 inhibitor lowers blood glucose and improves insulin sensitivity. Diabetes 2015, 64, 3396–3405. [Google Scholar]
- De Boer, J.F.; Dikkers, A.; Jurdzinski, A.; von Felden, J.; Gaestel, M.; Bavendiek, U.; Tietge, U.J. Mitogen-activated protein kinase-activated protein kinase 2 deficiency reduces insulin sensitivity in high-fat diet-fed mice. PLoS ONE 2014, 9, e106300. [Google Scholar]
- Scharf, M.; Neef, S.; Freund, R.; Geers-Knorr, C.; Franz-Wachtel, M.; Brandis, A.; Krone, D.; Schneider, H.; Groos, S.; Menon, M.B.; et al. Mitogen-activated protein kinase-activated protein kinases 2 and 3 regulate serca2a expression and fiber type composition to modulate skeletal muscle and cardiomyocyte function. Mol. Cell. Biol. 2013, 33, 2586–2602. [Google Scholar]
- Rajesh, M.; Batkai, S.; Kechrid, M.; Mukhopadhyay, P.; Lee, W.S.; Horvath, B.; Holovac, E.; Cinar, R.; Liaudet, L.; Mackie, K.; et al. Cannabinoid 1 receptor promotes cardiac dysfunction, oxidative stress, inflammation, and fibrosis in diabetic cardiomyopathy. Diabetes 2012, 61, 716–727. [Google Scholar]
- Wold, L.E.; Ceylan-Isik, A.F.; Fang, C.X.; Yang, X.; Li, S.Y.; Sreejayan, N.; Privratsky, J.R.; Ren, J. Metallothionein alleviates cardiac dysfunction in streptozotocin-induced diabetes: Role of Ca2+ cycling proteins, NADPH oxidase, poly(ADP-ribose) polymerase and myosin heavy chain isozyme. Free Radic. Biol. Med. 2006, 40, 1419–1429. [Google Scholar]
- Katz, M.G.; Fargnoli, A.S.; Williams, R.D.; Steuerwald, N.M.; Isidro, A.; Ivanina, A.V.; Sokolova, I.M.; Bridges, C.R. Safety and efficacy of high-dose adeno-associated virus 9 encoding sarcoplasmic reticulum Ca2+ adenosine triphosphatase delivered by molecular cardiac surgery with recirculating delivery in ovine ischemic cardiomyopathy. J. Thorac. Cardiovasc. Surg. 2014, 148, 1065–1072. [Google Scholar]
- Talukder, M.A.; Yang, F.; Nishijima, Y.; Chen, C.A.; Kalyanasundaram, A.; Periasamy, M.; Zweier, J.L. Reduced SERCA2a converts sub-lethal myocardial injury to infarction and affects postischemic functional recovery. J. Mol. Cell. Cardiol. 2009, 46, 285–287. [Google Scholar]
- Gurha, P.; Abreu-Goodger, C.; Wang, T.; Ramirez, M.O.; Drumond, A.L.; van Dongen, S.; Chen, Y.; Bartonicek, N.; Enright, A.J.; Lee, B.; et al. Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 2012, 125, 2751–2761. [Google Scholar]
- Park, J.K.; Ronkina, N.; Hoft, A.; Prohl, C.; Menne, J.; Gaestel, M.; Haller, H.; Meier, M. Deletion of MK2 signalling in vivo inhibits small HSP phosphorylation but not diabetic nephropathy. Nephrol. Dial. Transplant 2008, 23, 1844–1853. [Google Scholar]
- Oguiza, A.; Recio, C.; Lazaro, I.; Mallavia, B.; Blanco, J.; Egido, J.; Gomez-Guerrero, C. Peptide-based inhibition of IκB kinase/nuclear factor-κB pathway protects against diabetes-associated nephropathy and atherosclerosis in a mouse model of type 1 diabetes. Diabetologia 2015, 58, 1656–1667. [Google Scholar]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar]
- Latronico, M.V.; Condorelli, G. MicroRNAs and cardiac pathology. Nat. Rev. Cardiol. 2009, 6, 419–429. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of micrornas after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar]
- Montgomery, R.L.; Hullinger, T.G.; Semus, H.M.; Dickinson, B.A.; Seto, A.G.; Lynch, J.M.; Stack, C.; Latimer, P.A.; Olson, E.N.; van Rooij, E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation 2011, 124, 1537–1547. [Google Scholar]
- Grueter, C.E.; van Rooij, E.; Johnson, B.A.; DeLeon, S.M.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A cardiac microrna governs systemic energy homeostasis by regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar]
- Romaine, S.P.; Tomaszewski, M.; Condorelli, G.; Samani, N.J. Micrornas in cardiovascular disease: An introduction for clinicians. Heart 2015, 101, 921–928. [Google Scholar]
- Zhou, S.; Liu, Y.; Prater, K.; Zheng, Y.; Cai, L. Roles of microRNAs in pressure overload- and ischemia-related myocardial remodeling. Life Sci. 2013, 93, 855–862. [Google Scholar]
- Shen, E.; Diao, X.; Wang, X.; Chen, R.; Hu, B. MicroRNAs involved in the mitogen-activated protein kinase cascades pathway during glucose-induced cardiomyocyte hypertrophy. Am. J. Pathol. 2011, 179, 639–650. [Google Scholar]
- Tijsen, A.J.; van der Made, I.; van den Hoogenhof, M.M.; Wijnen, W.J.; van Deel, E.D.; de Groot, N.E.; Alekseev, S.; Fluiter, K.; Schroen, B.; Goumans, M.J.; et al. The microRNA-15 family inhibits the TGFβ-pathway in the heart. Cardiovasc. Res. 2014, 104, 61–71. [Google Scholar]
- Torella, D.; Ellison, G.M.; Torella, M.; Vicinanza, C.; Aquila, I.; Iaconetti, C.; Scalise, M.; Marino, F.; Henning, B.J.; Lewis, F.C.; et al. Carbonic anhydrase activation is associated with worsened pathological remodeling in human ischemic diabetic cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000434. [Google Scholar]
- Blumensatt, M.; Greulich, S.; Herzfeld de Wiza, D.; Mueller, H.; Maxhera, B.; Rabelink, M.J.; Hoeben, R.C.; Akhyari, P.; Al-Hasani, H.; Ruige, J.B.; et al. Activin A impairs insulin action in cardiomyocytes via up-regulation of mir-143. Cardiovasc. Res. 2013, 100, 201–210. [Google Scholar]
- Huang, B.; Qin, W.; Zhao, B.; Shi, Y.; Yao, C.; Li, J.; Xiao, H.; Jin, Y. MicroRNA expression profiling in diabetic gk rat model. Acta Biochim. Biophys. Sin. 2009, 41, 472–477. [Google Scholar]
- Liu, S.; Li, W.; Xu, M.; Huang, H.; Wang, J.; Chen, X. Micro-RNA 21targets dual specific phosphatase 8 to promote collagen synthesis in high glucose-treated primary cardiac fibroblasts. Can. J. Cardiol. 2014, 30, 1689–1699. [Google Scholar]
- Zhang, H.; Hao, Y.; Yang, J.; Zhou, Y.; Li, J.; Yin, S.; Sun, C.; Ma, M.; Huang, Y.; Xi, J.J. Genome-wide functional screening of mir-23b as a pleiotropic modulator suppressing cancer metastasis. Nat. Commun. 2011, 2, 554. [Google Scholar] [CrossRef]
- Zhu, S.; Pan, W.; Song, X.; Liu, Y.; Shao, X.; Tang, Y.; Liang, D.; He, D.; Wang, H.; Liu, W.; et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat. Med. 2012, 18, 1077–1086. [Google Scholar]
- He, J.; Li, Y.; Yang, X.; He, X.; Zhang, H.; He, J.; Zhang, L. The feedback regulation of pi3k-miR-19a, and MAPK-mir-23b/27b in endothelial cells under shear stress. Molecules 2012, 18, 1–13. [Google Scholar]
- Blumensatt, M.; Wronkowitz, N.; Wiza, C.; Cramer, A.; Mueller, H.; Rabelink, M.J.; Hoeben, R.C.; Eckel, J.; Sell, H.; Ouwens, D.M. Adipocyte-derived factors impair insulin signaling in differentiated human vascular smooth muscle cells via the upregulation of miR-143. Biochim. Biophys. Acta 2014, 1842, 275–283. [Google Scholar]
- Qian, L.; Van Laake, L.W.; Huang, Y.; Liu, S.; Wendland, M.F.; Srivastava, D. miR-24 inhibits apoptosis and represses BIM in mouse cardiomyocytes. J. Exp. Med. 2011, 208, 549–560. [Google Scholar]
- Zampetaki, A.; Kiechl, S.; Drozdov, I.; Willeit, P.; Mayr, U.; Prokopi, M.; Mayr, A.; Weger, S.; Oberhollenzer, F.; Bonora, E.; et al. Plasma microrna profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 2010, 107, 810–817. [Google Scholar]
- Xiang, Y. miR-24 in diabetes. Oncotarget 2015, 6, 16816–16817. [Google Scholar]
- Kiriakidou, M.; Nelson, P.T.; Kouranov, A.; Fitziev, P.; Bouyioukos, C.; Mourelatos, Z.; Hatzigeorgiou, A. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004, 18, 1165–1178. [Google Scholar]
- Huang, W.; Tian, S.S.; Hang, P.Z.; Sun, C.; Guo, J.; Du, Z.M. Combination of microRNA-21 and microRNA-146a attenuates cardiac dysfunction and apoptosis during acute myocardial infarction in mice. Mol. Ther. Nucleic Acids 2016, 5, e296. [Google Scholar]
- Zaman, M.S.; Shahryari, V.; Deng, G.; Thamminana, S.; Saini, S.; Majid, S.; Chang, I.; Hirata, H.; Ueno, K.; Yamamura, S.; et al. Up-regulation of microRNA-21 correlates with lower kidney cancer survival. PLoS ONE 2012, 7, e31060. [Google Scholar]


{kind=link}
{kind=link}
| Inhibitors | Isoforms | Model | Response | References |
|---|---|---|---|---|
| SB203580 | α, β | Multiple injections of STZ (50 mg/kg i.p. for five days) in C57/BL6 mice | Improved cardiac function | [15] |
| SB202190 | α, β | Single injection of STZ (65 mg/kg i.p.) in rats | Prevented cardiomyocyte apoptosis | [60] |
| PD169316 | α, β | Human adult ventricular cardiomyocytes treated with palmitate | Increased vasorelaxation | [4] |
| BIRB 0796 | α, β, γ and, δ | Cardiac myofibroblasts treated with 10 ng/mL of IL-1α for six hours | Reduced inflammatory cytokine release | [61] |
| MicroRNA | Location | Model | Response | References |
|---|---|---|---|---|
| miR-373 | Downstream | Single injection of STZ (150 mg/kg i.p.) in C57/BL6 mice | Prevent cardiomyocyte hypertrophy | [88] |
| miR-23b | Downstream | LV of T2D patients and cardiomyocytes from rat high glucose-induced model | Prevent cardiomyocyte hypertrophy | [90] |
| miR-143 | Downstream | Primary rat cardiomyocytes exposed to adipose tissue from T2D patients | Increase cardiomyocyte insulin resistance | [91] |
| miR-24 | Upstream | T2D patients and Goto-Kakizaki (GK) rat | Prevent cardiomyocyte apoptosis | [92] |
| miR-21 | Upstream | Rat cardiac fibroblasts with high glucose treatment (in vitro) | Prevent cardiac fibrosis | [93] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Ding, L.; Ji, H.; Xu, Z.; Liu, Q.; Zheng, Y. The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2016, 17, 1037. https://doi.org/10.3390/ijms17071037
Wang S, Ding L, Ji H, Xu Z, Liu Q, Zheng Y. The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy. International Journal of Molecular Sciences. 2016; 17(7):1037. https://doi.org/10.3390/ijms17071037
Chicago/Turabian StyleWang, Shudong, Lijuan Ding, Honglei Ji, Zheng Xu, Quan Liu, and Yang Zheng. 2016. "The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy" International Journal of Molecular Sciences 17, no. 7: 1037. https://doi.org/10.3390/ijms17071037
APA StyleWang, S., Ding, L., Ji, H., Xu, Z., Liu, Q., & Zheng, Y. (2016). The Role of p38 MAPK in the Development of Diabetic Cardiomyopathy. International Journal of Molecular Sciences, 17(7), 1037. https://doi.org/10.3390/ijms17071037