
Homocysteine Aggravates Cortical Neural Cell Injury through Neuronal Autophagy Overactivation following Rat Cerebral Ischemia-Reperfusion

Abstract
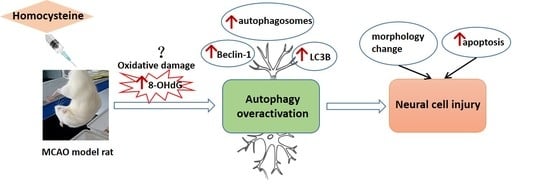
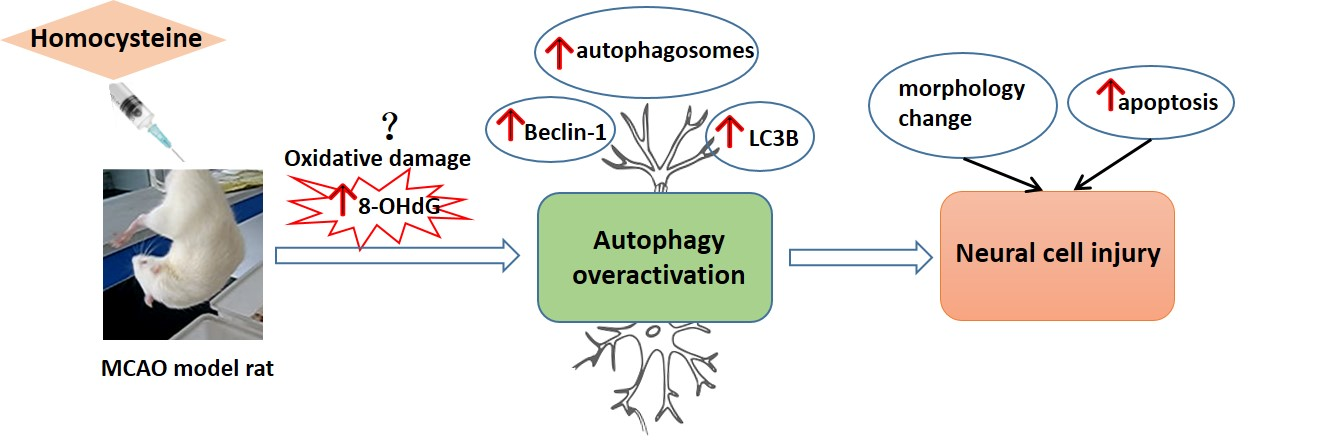
:
1. Introduction
2. Results
2.1. Serum Hcy Concentration of Rats
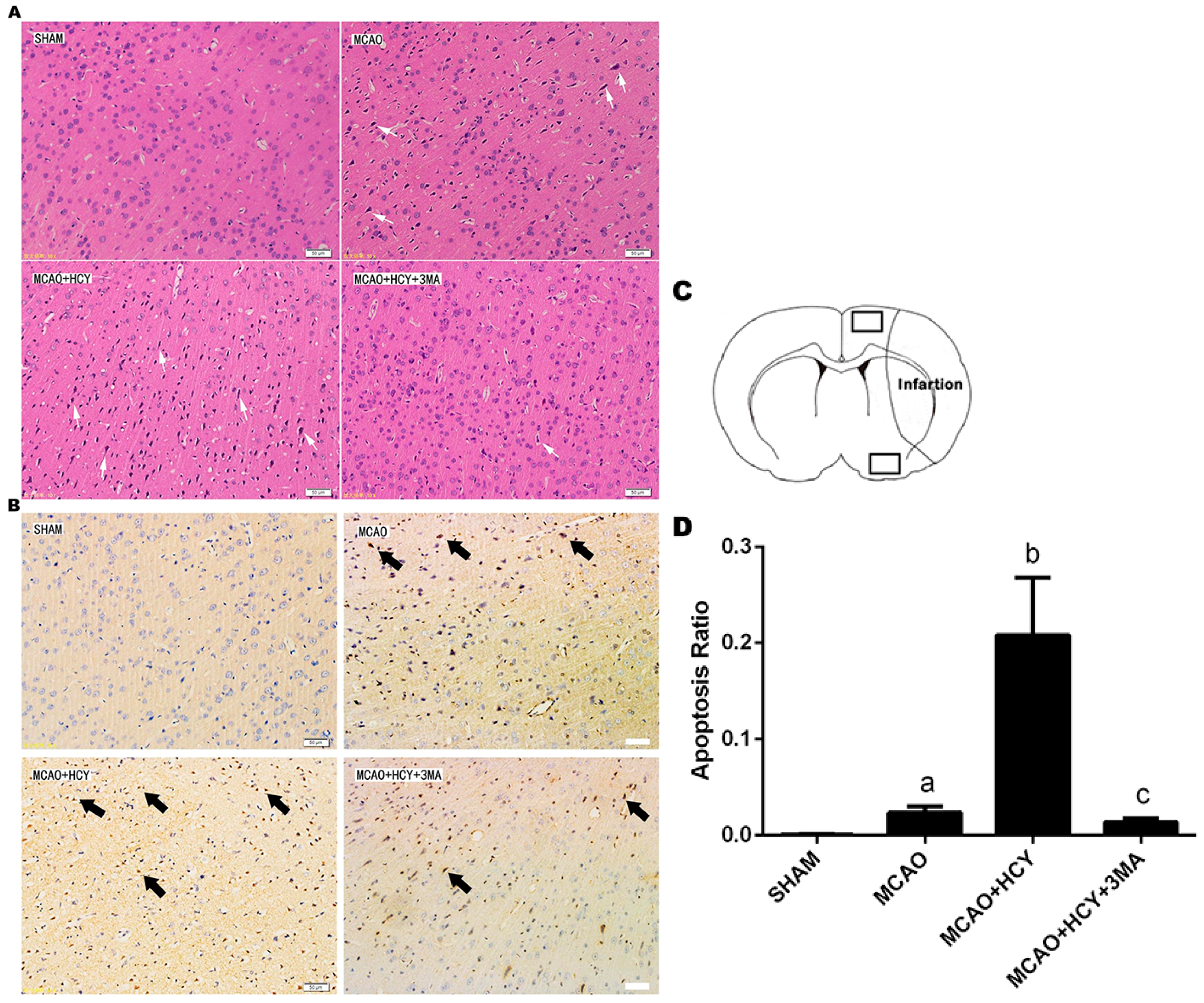
2.2. Hcy-Induced Neural Cell Injury in Ischemia Brain by Observation of Pathological Morphology and Apoptosis
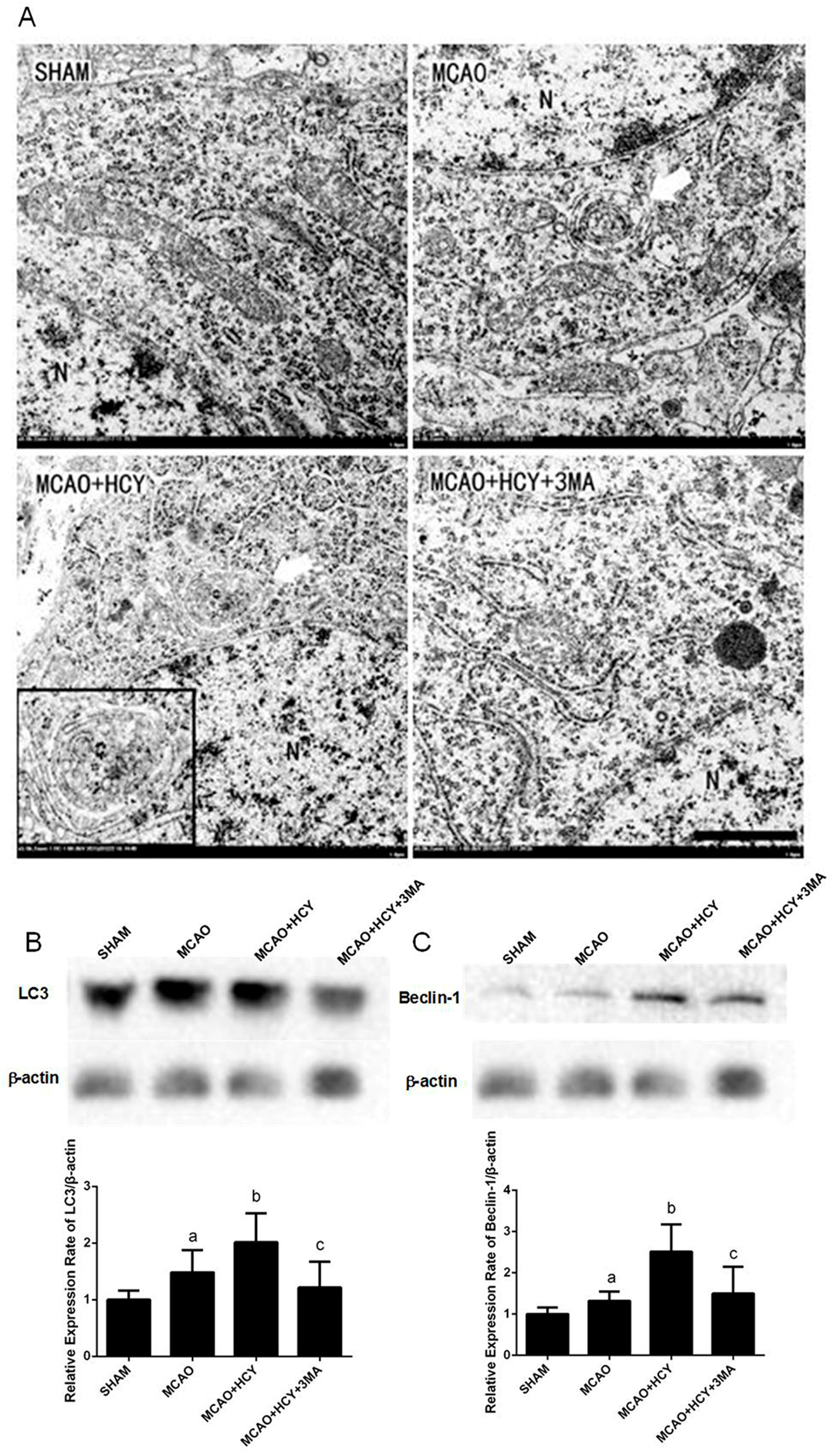
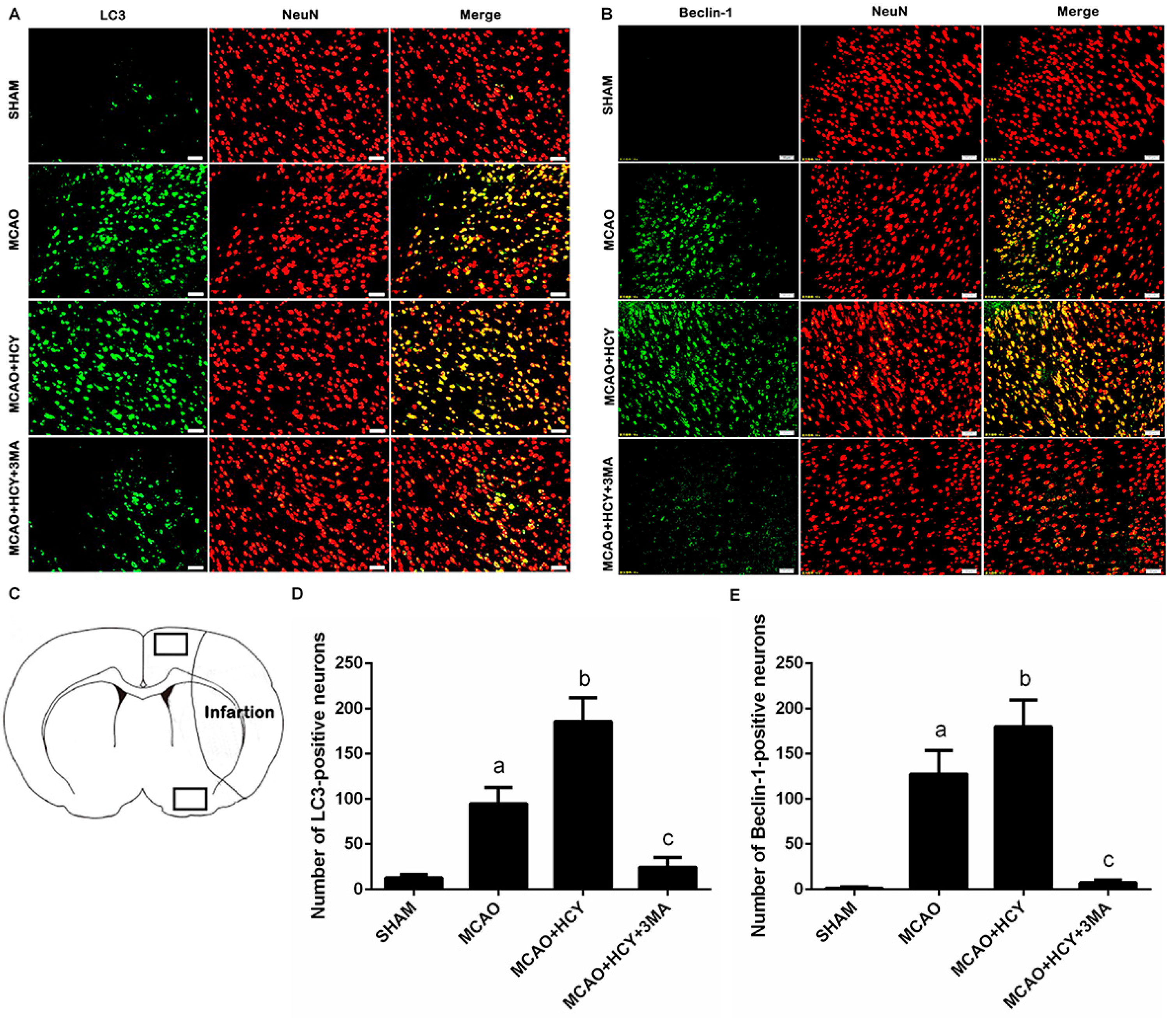
2.3. Hcy Induced Autophagosomes Accumulation and Protein Expression of LC3B and Beclin-1 in Cortex Neurons Following MCAO Injury
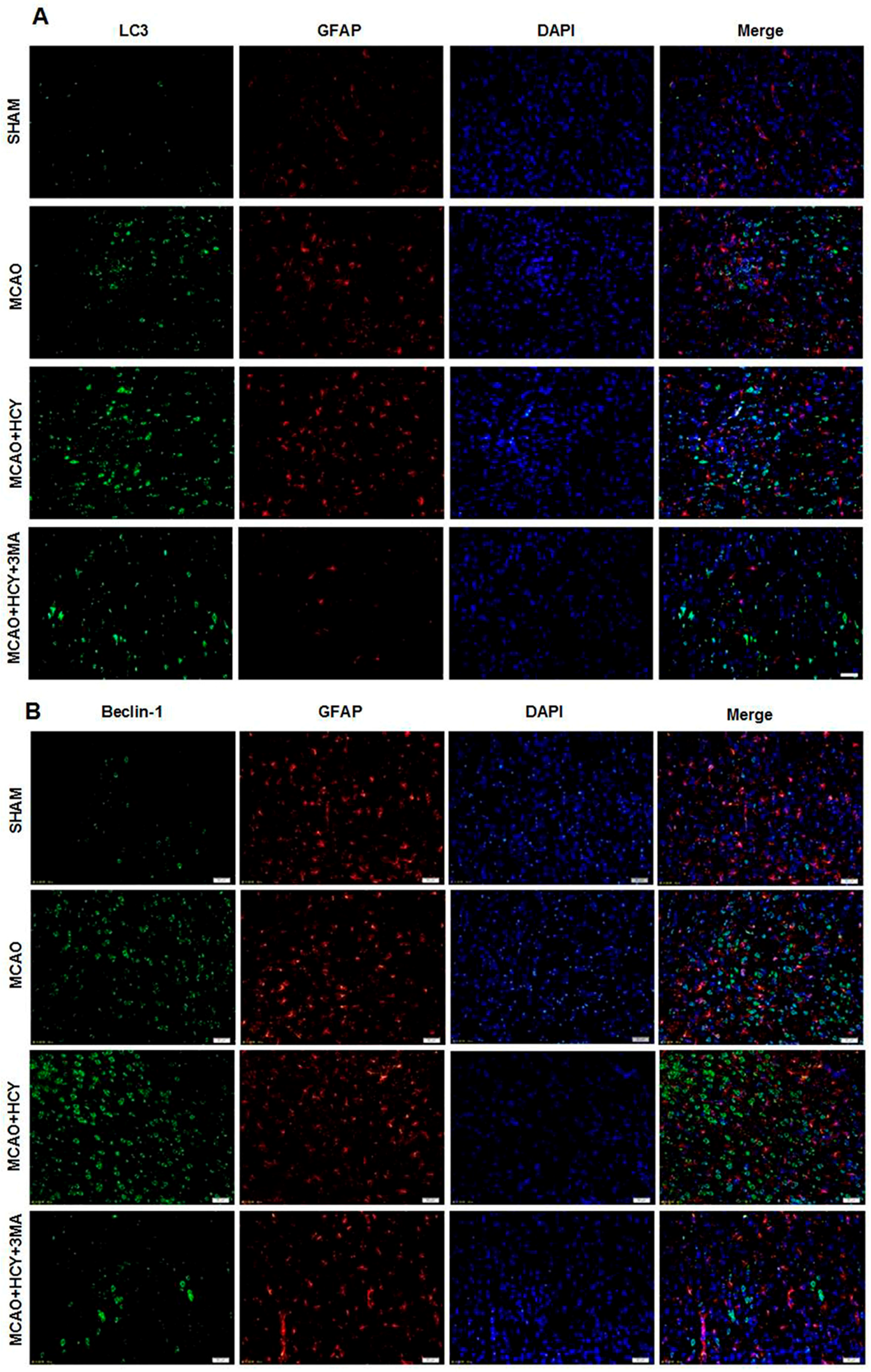
2.4. Hcy Induced LC3B and Beclin-1 Protein Accumulation in Neurons Not Astrocytes Following Ischemia by Immunofluorescence
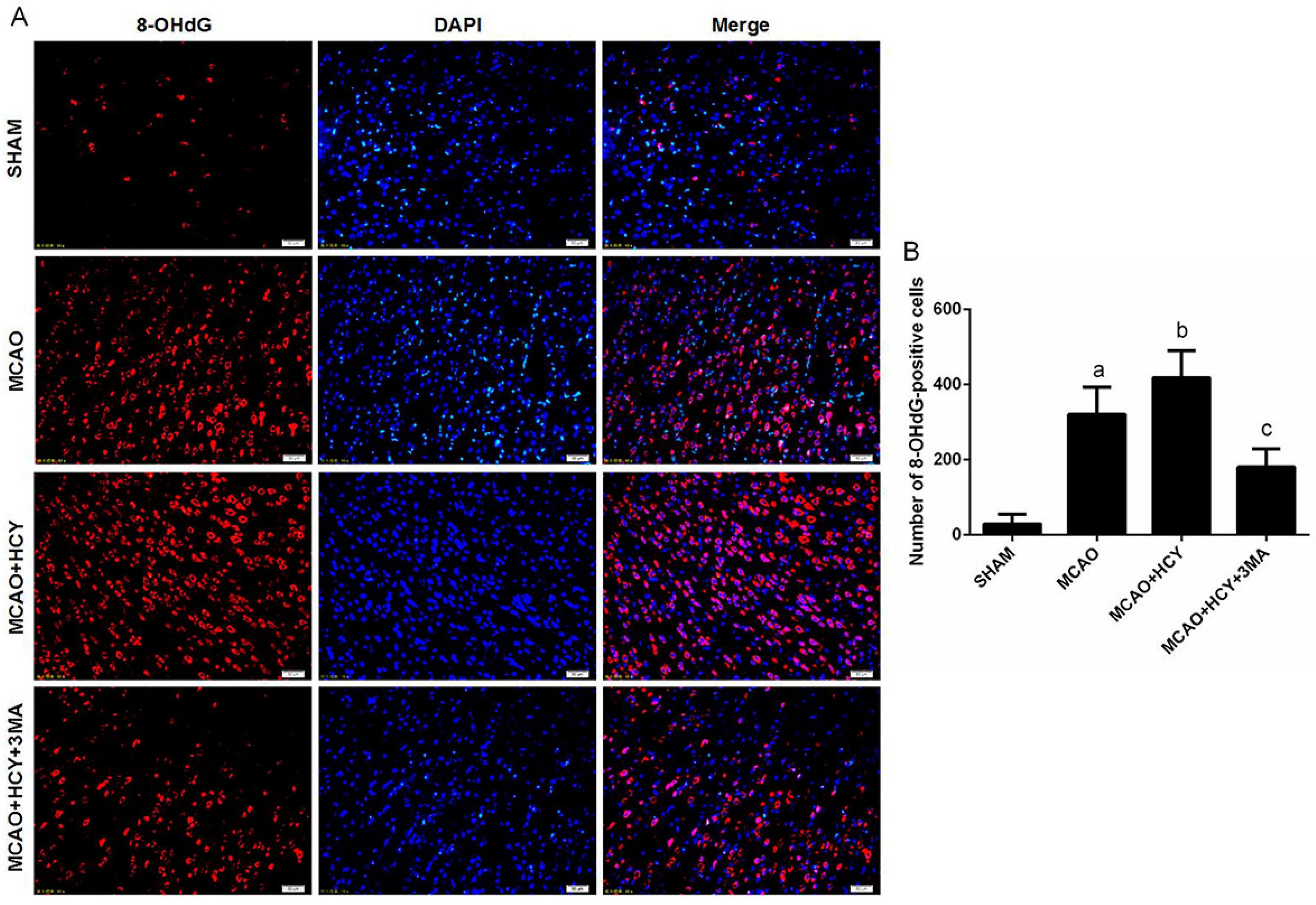
2.5. Hcy Increased 8-OHdG Protein Expression Following Ischemia
3. Discussion
4. Materials and Methods
4.1. Experimental Design
4.2. Surgical Procedures
4.3. Serum Homocysteine Concentration
4.4. Transmission Electron Microscopy (TEM)
4.5. Preparation of Paraffin Section
4.6. HE and TUNEL Staining
4.7. Immunofluorescence
4.8. Western Blot
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yang, Y.; Gao, K.; Hu, Z.; Li, W.; Davies, H.; Ling, S.; Rudd, J.A.; Fang, M. Autophagy upregulation and apoptosis downregulation in DAHP and triptolide treated cerebral ischemia. Mediat. Inflamm. 2015, 2015, 120–198. [Google Scholar] [CrossRef] [PubMed]
- Savage, S.; Ma, D. The neurotoxicity of nitrous oxide: The facts and “putative” mechanisms. Brain Sci. 2014, 4, 73–90. [Google Scholar] [CrossRef] [PubMed]
- Bostom, A.G.; Rosenberg, I.H.; Silbershatz, H.; Jacques, P.F.; Selhub, J.; D’Agostino, R.B.; Wilson, P.W.; Wolf, P.A. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: The Framingham Study. Ann. Intern. Med. 1999, 131, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Chen, Y.M.; Yao, G.E.; Yao, C.S.; Zhao, H.M.; Jia, X.D.; Zhang, Y.Y.; Ge, J.L.; Qiu, E.C.; Ding, C.Y. Effects of differences in serum total homocysteine, folate, and vitamin B12 on cognitive impairment in stroke patients. BMC Neurol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Waeckel, L.; Bignon, J.; Liu, J.M.; Markovits, D.; Ebrahimian, T.G.; Vilar, J.; Mees, B.; Blanc-Brude, O.; Barateau, V.; Le Ricousse-Roussanne, S.; et al. Tetrapeptide AcSDKP induces postischemic neovascularization through monocyte chemoattractant protein-1 signaling. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [PubMed]
- Ginet, V.; Spiehlmann, A.; Rummel, C.; Rudinskiy, N.; Grishchuk, Y.; Luthi-Carter, R.; Clarke, P.G.; Truttmann, A.C.; Puyal, J. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy 2014, 10, 846–860. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Weng, J.; Zhao, L.; Li, X.M.; Gao, T.M.; Kong, J. Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci. Ther. 2012, 18, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Guidi, I.; Galimberti, D.; Lonati, S.; Novembrino, C.; Bamonti, F.; Tiriticco, M.; Fenoglio, C.; Venturelli, E.; Baron, P.; Bresolin, N. Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol. Aging 2006, 27, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Vacek, J.C.; Givvimani, S.; Sen, U.; Tyagi, S.C. Cardiac specific deletion of N-methyl-d-aspartate receptor 1 ameliorates mtMMP-9 mediated autophagy/mitophagy in hyperhomocysteinemia. J. Recept. Signal Transduct. Res. 2010, 30, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Malishkevich, A.; Shemer, Z.; Udawela, M.; Gibbons, A.; Scarr, E.; Dean, B.; Levine, J.; Agam, G.; Gozes, I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 2015, 20, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Nitatori, T.; Sato, N.; Waguri, S.; Karasawa, Y.; Araki, H.; Shibanai, K.; Kominami, E.; Uchiyama, Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischemia is apoptosis. J. Neurosci. 1995, 15, 1001–1011. [Google Scholar] [PubMed]
- Adhami, F.; Schloemer, A.; Kuan, C.K. The roles of autophagy in cerebral ischemia. Autophagy 2007, 3, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhang, H.; Bai, X.; Lu, Y.; Dong, H.; Xiong, L. Autophagy activation is involved in neuroprotection induced by hyperbaric oxygen preconditioning against focal cerebral ischemia in rats. Brain Res. 2011, 1402, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Zhang, Y.; Li, J.; Zhang, J.; Li, Y.; Dang, C.; Li, C.; Fan, Y.; Yu, J.; Pei, Z.; et al. Beclin 1 knockdown inhibits autophagic activation and prevents the secondary neurodegenerative damage in the ipsilateral thalamus following focal cerebral infarction. Autophagy 2012, 8, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, H.; Yuan, Y.; Gao, J.; Shen, Z.; Cheng, Y.; Shen, Y.; Wang, R.R.; Wang, X.; Hu, W.W.; et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013, 9, 1321–1333. [Google Scholar] [CrossRef] [PubMed]
- Adhami, F.; Liao, G.; Morozov, Y.M.; Schloemer, A.; Schmithorst, V.J.; Lorenz, J.N.; Dunn, R.S.; Vorhees, C.V.; Wills-Karp, M.; Degen, J.L.; et al. Cerebral ischemia-hypoxia induces intravascular coagulation and autophagy. Am. J. Pathol. 2006, 169, 566–583. [Google Scholar] [CrossRef] [PubMed]
- Rami, A.; Langhagen, A.; Steiger, S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol. Dis. 2008, 29, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Choubey, V.; Cagalinec, M.; Liiv, J.; Safiulina, D.; Hickey, M.A.; Kuum, M.; Liiv, M.; Anwar, T.; Eskelinen, E.L.; Kaasik, A. BECN1 is involved in the initiation of mitophagy: It facilitates PARK2 translocation to mitochondria. Autophagy 2014, 10, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Sun, L.; Wang, C.; Huang, H.; Liu, J.; Liao, W.; Shi, M. LC3A-positive “stone-like” structures predict an adverse prognosis of gastric cancer. Anat. Rec. 2014, 297, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Wang, Z.; Wang, L.; Zhang, L.; Xiang, X.; Wang, Z.; Chong, T. Lower mRNA and protein expression levels of LC3 and Beclin1, markers of autophagy, were correlated with progression of renal clear cell carcinoma. Jpn. J. Clin. Oncol. 2013, 43, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.D.; Sheng, R.; Zhang, L.S.; Han, R.; Zhang, X.; Zhang, X.D.; Han, F.; Fukunaga, K.; Qin, Z.H. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 2008, 4, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Jiang, Y.; Zhou, Z.; Yue, X.; Wei, N.; Chen, Z.; Ma, M.; Xu, G.; Liu, X. Intranasal ginsenoside Rb1 targets the brain and ameliorates cerebral ischemia/reperfusion injury in rats. Biol. Pharm. Bull. 2011, 34, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Li, J.; Ni, W.; Shen, Y.W.; Zhang, X.P. Peroxisome proliferator-activated receptor-γ agonist 15d-prostaglandin J2 mediates neuronal autophagy after cerebral ischemia-reperfusion injury. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Hsiao, T.Y.; Chien, C.T.; Lai, M.K. Ischemic conditioning by short periods of reperfusion attenuates renal ischemia/reperfusion induced apoptosis and autophagy in the rat. J. Biomed. Sci. 2009, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of autophagy by reactive oxygen species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.N.; Viarengo, A.; Donkin, P.; Hawkins, A.J. Autophagic and lysosomal reactions to stress in the hepatopancreas of blue mussels. Aquat. Toxicol. 2007, 84, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Harris, V.A.; Kumar, S.; Mansour, H.M.; Black, S.M. Autophagy in neonatal hypoxia ischemic brain is associated with oxidative stress. Redox Biol. 2015, 6, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar]
- Ho, P.I.; Ashline, D.; Dhitavat, S.; Ortiz, D.; Collins, S.C.; Shea, T.B.; Rogers, E. Folate deprivation induces neurodegeneration: Roles of oxidative stress and increased homocysteine. Neurobiol. Dis. 2003, 14, 32–42. [Google Scholar] [CrossRef]
- Tyagi, N.; Moshal, K.S.; Sen, U.; Vacek, T.P.; Kumar, M.; Hughes, W.M., Jr.; Kundu, S.; Tyagi, S.C. H2S protects against methionine-induced oxidative stress in brain endothelial cells. Antioxid. Redox Signal. 2009, 11, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Plachetka, A.; Adamek, B.; Strzelczyk, JK.; Krakowczyk, Ł.; Migula, P.; Nowak, P.; Wiczkowski, A. 8-hydroxy-2′-deoxyguanosine in colorectal adenocarcinoma—Is it a result of oxidative stress? Med. Sci. Monit. 2013, 19, 690–695. [Google Scholar] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Gu, C.; Ma, M.; Cong, Q.; Guo, T.; Ma, D.; Li, B. A mouse model of premature ovarian insufficiency induced by tripterygium glycoside via subcutaneous injection. Int. J. Clin. Exp. Pathol. 2014, 7, 144–151. [Google Scholar] [PubMed]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Before Intervention (μM) | After Intervention (μM) |
|---|---|---|
| SHAM | 5.40 ± 2.72 | 5.20 ± 2.02 |
| MCAO | 7.20 ± 0.80 | 6.90 ± 2.47 |
| MCAO + HCY | 7.00 ± 1.77 | 15.07 ± 2.66 a,b,* |
| MCAO + HCY + 3MA | 6.40 ± 2.26 | 10.53±0.83 a,b,* |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Huang, G.; Chen, S.; Gou, Y.; Dong, Z.; Zhang, X. Homocysteine Aggravates Cortical Neural Cell Injury through Neuronal Autophagy Overactivation following Rat Cerebral Ischemia-Reperfusion. Int. J. Mol. Sci. 2016, 17, 1196. https://doi.org/10.3390/ijms17081196
Zhao Y, Huang G, Chen S, Gou Y, Dong Z, Zhang X. Homocysteine Aggravates Cortical Neural Cell Injury through Neuronal Autophagy Overactivation following Rat Cerebral Ischemia-Reperfusion. International Journal of Molecular Sciences. 2016; 17(8):1196. https://doi.org/10.3390/ijms17081196
Chicago/Turabian StyleZhao, Yaqian, Guowei Huang, Shuang Chen, Yun Gou, Zhiping Dong, and Xumei Zhang. 2016. "Homocysteine Aggravates Cortical Neural Cell Injury through Neuronal Autophagy Overactivation following Rat Cerebral Ischemia-Reperfusion" International Journal of Molecular Sciences 17, no. 8: 1196. https://doi.org/10.3390/ijms17081196
APA StyleZhao, Y., Huang, G., Chen, S., Gou, Y., Dong, Z., & Zhang, X. (2016). Homocysteine Aggravates Cortical Neural Cell Injury through Neuronal Autophagy Overactivation following Rat Cerebral Ischemia-Reperfusion. International Journal of Molecular Sciences, 17(8), 1196. https://doi.org/10.3390/ijms17081196