
The Role of Cyclo(His-Pro) in Neurodegeneration

 ,
, 
Abstract
:
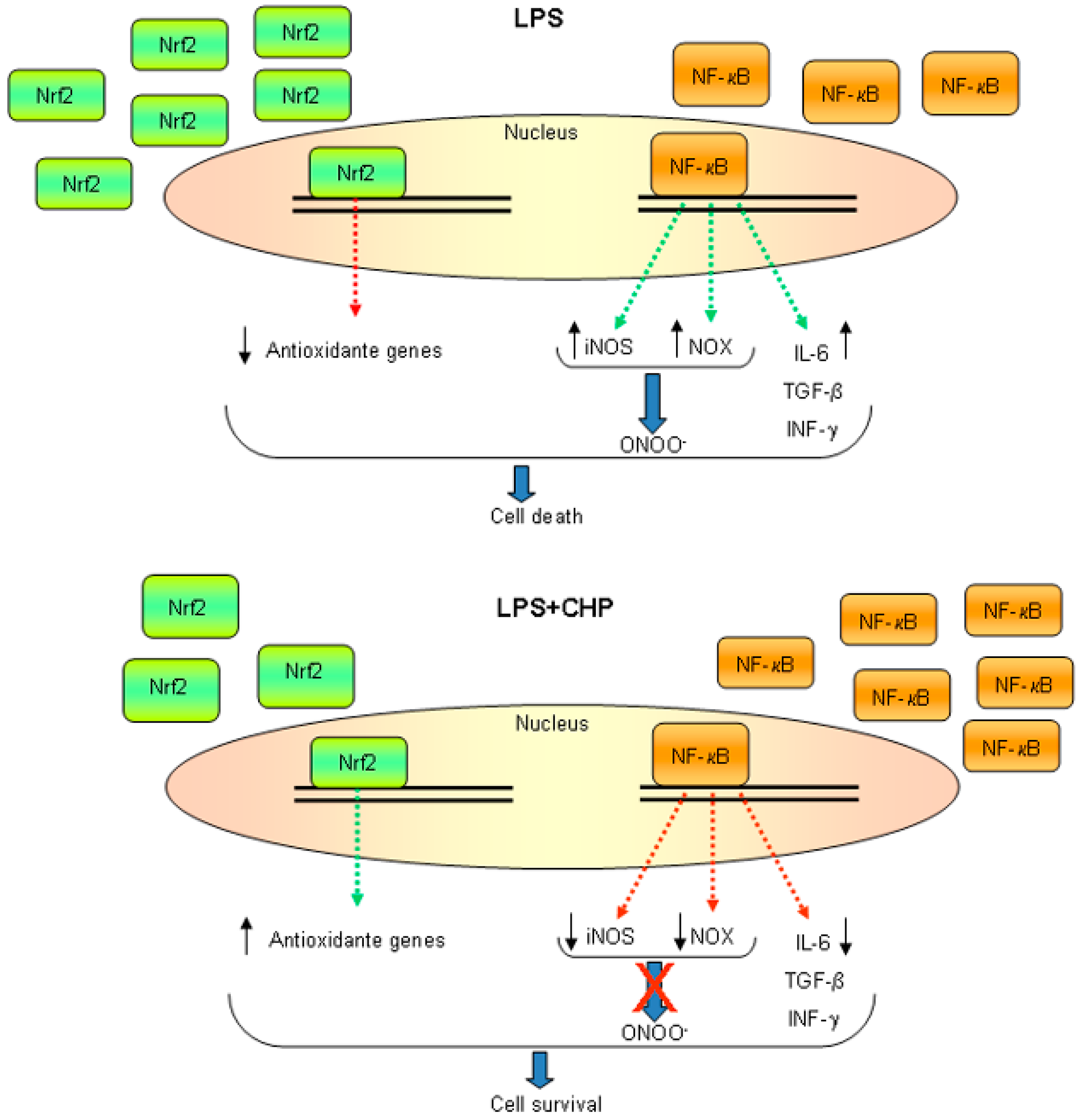
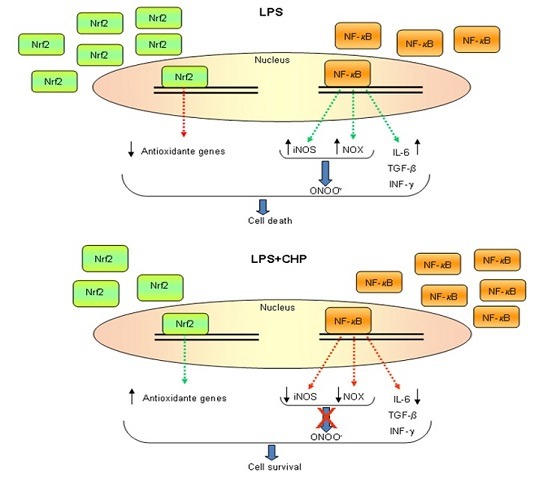{kind=link}
{kind=link}
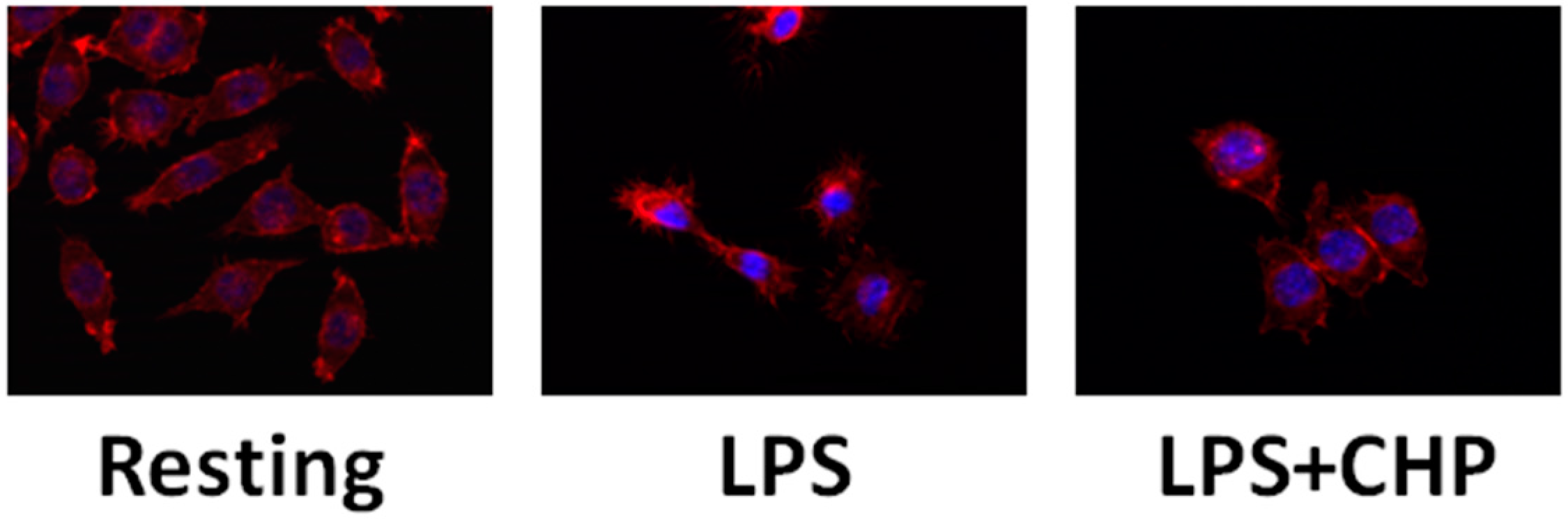{kind=link}
{kind=link}
{kind=link}
1. Introduction
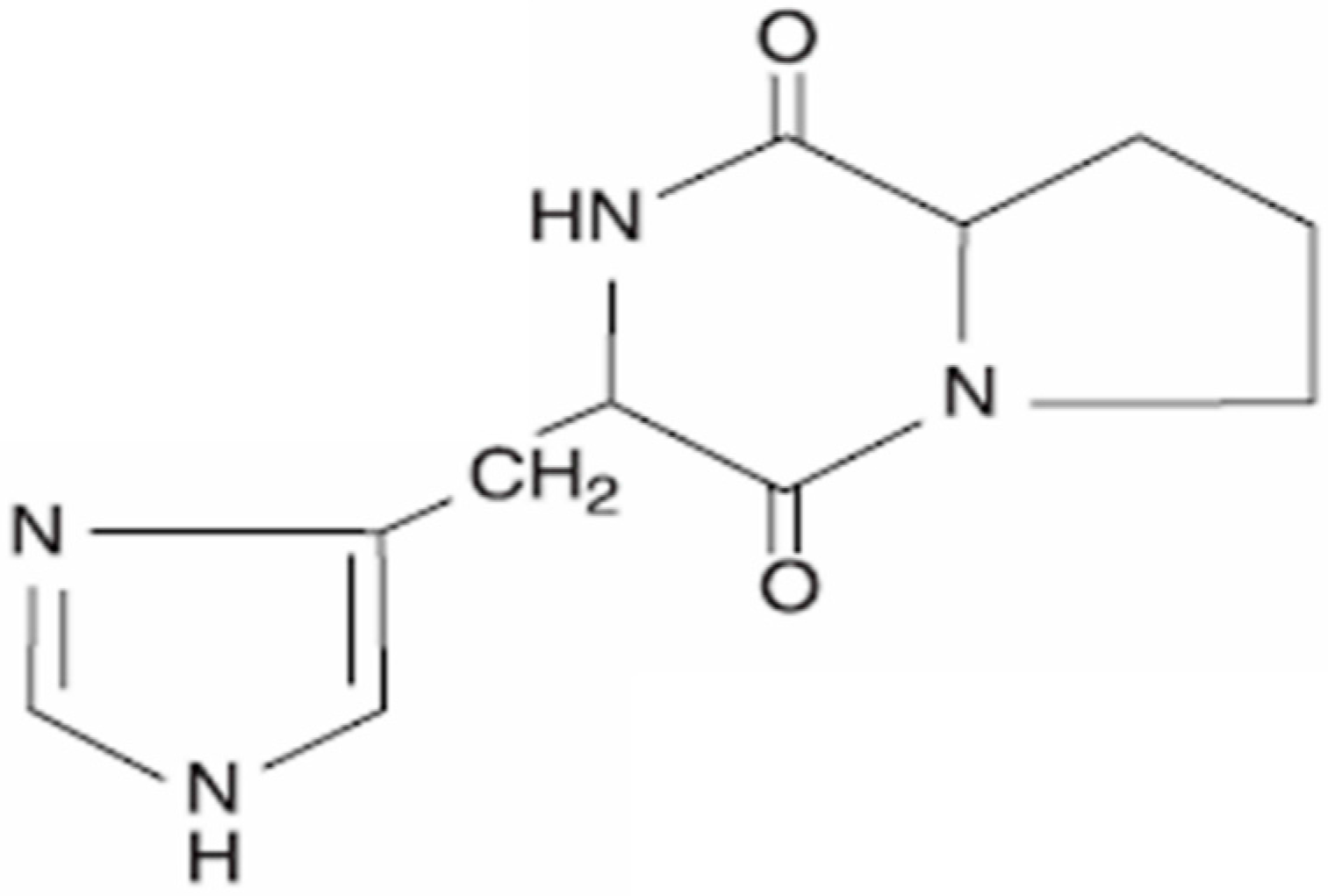
2. Cyclo(His-Pro): Chemistry and Biology
Cyclo(His-Pro) as a Key Substrate of Organic Cation Transporter
3. Role of Cyclo(His-Pro) in Common Mechanisms of Neurodegeneration
3.1. Oxidative and Nitrosative Stress
3.2. Endoplasmic Reticulum Stress
3.3. Excitotoxicity and Calcium Overload
4. Neuroinflammation
4.1. Cyclo(His-Pro) Acts as an Anti-Inflammatory Agent
4.2. Cyclo(His-Pro) Reduces Microgliosis/Neuroinflammation
5. Role of Cyclo(His-Pro) in in Vitro Models of Familial Amyotrophic Lateral Sclerosis
6. Conclusions
Author Contributions
Conflicts of Interest
References
- Faden, A.I. Pharmacological treatment of central nervous system trauma. Pharmacol. Toxicol. 1996, 78, 12–17. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Stoica, B. Neuroprotection: Challenges and opportunities. Arch. Neurol. 2007, 64, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Knoblach, S.M.; Cernak, I.; Fan, L.; Vink, R.; Araldi, G.L.; Fricke, S.T.; Roth, B.L.; Kozikowski, A.P. Noveldiketopiperazine enhances motor and cognitive recovery after traumatic brain injury in rats and shows neuroprotection in vitro and in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Knoblach, S.M.; Movsesyan, V.A.; Cernak, I. Novel small peptides with neuroprotective and nootropic properties. J. Alzheimers Dis. 2004, 6, S93–S97. [Google Scholar] [PubMed]
- Faden, A.I.; Knoblach, S.M.; Movsesyan, V.A.; Lea, P.M.; Cernak, I. Novel neuroprotective tripeptides and dipeptides. Ann. N. Y. Acad. Sci. 2005, 1053, 472–481. [Google Scholar] [CrossRef]
- Loane, D.J.; Faden, A.I. Neuroprotection for traumatic brain injury: Translational challenges and emerging therapeutic strategies. Trends Pharmacol. Sci. 2010, 31, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Bellezza, I.; Grottelli, S.; Pinnen, F.; Brunetti, L.; Vacca, M. Phosphoproteomic analysis of the effect of cyclo-[His-Pro] dipeptide on PC12 cells. Peptides 2006, 27, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Bellezza, I.; Grottelli, S.; Galli, F. Focus on cyclo(His-Pro): History and perspectives as antioxidant peptide. Amino Acids 2008, 35, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Conte, C.; Grottelli, S.; Bellezza, I.; Cacciatore, I.; Bolaños, J.P. Cyclo(His-Pro) promotes cytoprotection by activating Nrf2-mediated up-regulation of antioxidant defence. J. Cell. Mol. Med. 2009, 13, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Conte, C.; Grottelli, S.; Bellezza, I.; Emiliani, C.; Bolaños, J.P. Cyclo(His-Pro) up-regulates heme oxygenase 1 via activation of Nrf2-ARE signalling. J. Neurochem. 2009, 111, 956–966. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Faden, A.I.; Araldi, G.L. Cyclic Dipeptides and Azetidinone Compounds and Their Use in Treating CNS Injury and Neurodegenerative Disorders. U.S. Patent 7,202,279, 10 April 2007. [Google Scholar]
- Bellezza, I.; Peirce, M.J.; Minelli, A. Cyclic dipeptides: From bugs to brain. Trends Mol. Med. 2014, 20, 551–558. [Google Scholar] [CrossRef] [PubMed]
- Prakash, K.R.; Tang, Y.; Kozikowski, A.P.; Flippen-Anderson, J.L.; Knoblach, S.M.; Faden, A.I. Synthesis and biological activity of novel neuroprotective diketopiperazines. Bioorg. Med. Chem. 2002, 10, 3043–3048. [Google Scholar] [CrossRef]
- Faden, A.I. TRH analog YM-14673 improves outcome following traumatic brain and spinal cord injury in rats: Dose-response studies. Brain Res. 1989, 486, 228–235. [Google Scholar] [CrossRef]
- Faden, A.I.; Fox, G.B.; Fan, L.; Araldi, G.L.; Qiao, L.; Wang, S.; Kozikowski, A.P. Novel TRH analog improves motor and cognitive recovery after traumatic brain injury in rodents. Am. J. Physiol. 1999, 277, R1196–R1204. [Google Scholar] [PubMed]
- Ao, Y.; Toy, N.; Song, M.K.; Go, V.L.; Yang, H. Altered glucose and insulin responses to brain medullary thyrotropin-releasing hormone (TRH)-induced autonomic activation in type 2 diabetic Goto-Kakizaki rats. Endocrinology 2005, 146, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Scharfmann, R.; Morgat, J.L.; Aratan-Spire, S. Presence of a particulate thyrotropin-releasing hormone-degrading pyroglutamate aminopeptidase activity in rat liver. Neuroendocrinology 1989, 49, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.L.; Richardson, K.S.; Hansen, S.; Friesen, A.J. Identification of the diketopiperazine of histidylproline in human urine. J. Biol. Chem. 1965, 240, 4540–4542. [Google Scholar] [PubMed]
- Mizuma, T.; Masubuchi, S.; Awazu, S. Intestinal absorption of stable cyclic glycylphenylalanine: Comparison with the linear form. J. Pharm. Pharmacol. 1997, 49, 1067–1071. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Jaspan, J.B. Orally administered cyclo(His-Pro) reduces ethanol-induced narcosis in mice. Pharmacol. Biochem. Behav. 1992, 43, 939–941. [Google Scholar] [CrossRef]
- Banks, W.A.; Kastin, A.J.; Akerstrom, V.; Jaspan, J.B. Radioactively iodinated cyclo(His-Pro) crosses the blood-brain barrier and reverses ethanol-induced narcosis. Am. J. Physiol. 1993, 264, E723–E729. [Google Scholar] [PubMed]
- Jaspan, J.B.; Banks, W.A.; Kastin, A.J. Study of passage of peptides across the blood-brain barrier: Biological effects of cyclo(His-Pro) after intravenous and oral administration. Ann. N. Y. Acad. Sci. 1994, 739, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C. Limited proteolysis and physiological regulation: An example from thyrotropin-releasing hormone metabolism. Thyroid 1998, 8, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Prasad, C. Bioactive cyclic dipeptides. Peptides 1995, 16, 1511–1564. [Google Scholar] [CrossRef]
- Hilton, C.W.; Prasad, C.; Svec, F.; Vo, P.; Reddy, S. Cyclo(His-Pro) in nutritional supplements. Lancet 1990, 336, 1455. [Google Scholar] [CrossRef]
- Mizuma, H.; Legardeur, B.Y.; Prasad, C.; Hilton, C.W. The bioactive peptide cyclo(His-Pro) may be absorbed following ingestion of nutritional supplements that contain it. J. Am. Coll. Nutr. 1996, 15, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Uyemura, K.; Dhanani, S.; Yamaguchi, D.T.; Song, M.K. Metabolism and toxicity of high doses of cyclo(His-Pro) plus zinc in healthy human subjects. J. Drug Metab. Toxicol. 2010, 1, 105. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Deliv. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Karpowich, N.K.; Song, J.M.; Cocco, N.; Wang, D.N. ATP binding drives substrate capture in an ECF transporter by a release-and-catch mechanism. Nat. Struct. Mol. Biol. 2015, 22, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Daniel, H.; Kottra, G. The proton oligopeptide cotransporter family SLC15 in physiology and pharmacology. Pflügers Arch. 2004, 447, 610–618. [Google Scholar] [PubMed]
- Hagenbuch, B.; Meier, P.J. Organic anion transporting polypeptides of the OATP/SLC21 family: Phylogenetic classification as OATP/ SLCO superfamily, new nomenclature and molecular/functional properties. Pflügers Arch. 2004, 447, 653–665. [Google Scholar] [CrossRef] [PubMed]
- van Montfoort, J.E.; Hagenbuch, B.; Groothuis, G.M.; Koepsell, H.; Meier, P.J.; Meijer, D.K. Drug uptake systems in liver and kidney. Curr. Drug Metab. 2003, 4, 185–211. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H.; Endou, H. The SLC22 drug transporter family. Pflügers Arch. 2004, 447, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Matsumoto, T.; Morimoto, R.; Arioka, S.; Omote, H.; Moriyama, Y. A human transporter protein that mediates the final excretion step for toxic organic cations. Proc. Natl. Acad. Sci. USA 2005, 102, 17923–17928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelis, R.M.; Wright, S.H. SLC22, SLC44, and SLC47 Transporters-organic anion and cation transporters: Molecular and cellular properties. Curr. Top. Membr. 2014, 73, 233–261. [Google Scholar] [PubMed]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific organic cation transporters: Structure, function, physiological roles, and biopharmaceutical implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef] [PubMed]
- Taubert, D.; Grimberg, G.; Stenzel, W.; Schömig, E. Identification of the endogenous key substrates of the human organic cation transporter OCT2 and their implication in function of dopaminergic neurons. PLoS ONE 2007, 2, e3852. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. The SLC22 family with transporters of organic cations, anions and zwitterions. Mol. Asp. Med. 2013, 34, 413–435. [Google Scholar] [CrossRef] [PubMed]
- Hacker, K.; Maas, R.; Kornhuber, J.; Fromm, M.F.; Zolk, O. Substrate-dependent inhibition of the human organic cation Transporter OCT2: A comparison of metformin with experimental substrates. PLoS ONE 2015, 10, e0136451. [Google Scholar]
- Ikegami, H.; Prasad, C. Neuropeptide-dopamine interactions. V. Cyclo(His-Pro) regulation of striatal dopamine transporter complex. Peptides 1990, 11, 145–148. [Google Scholar] [CrossRef]
- Faden, A.I.; Movsesyan, V.A.; Knoblach, S.M.; Ahmed, F.; Cernak, I. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology 2005, 49, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Stoica, B.; Byrnes, K.; Faden, A.I. Multifunctional drug treatment in neurotrauma. Neurotherapeutics 2009, 6, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Laurie, C.; Mosley, R.L.; Gendelman, H.E. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 297–325. [Google Scholar] [PubMed]
- Hsu, M.; Srinivas, B.; Kumar, J.; Subramanian, R.; Andersen, J. Glutathione depletion resulting in selective mitochon-drial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: Implications for Parkinson’s disease. J. Neurochem. 2005, 92, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swansonet, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Dasuri, K.; Zhang, L.; Keller, J.N. Oxidative stress, neurodegeneration, and the balance of protein degradation and protein synthesis. Free Radic. Biol. Med. 2013, 62, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Contestabile, A.; Monti, B.; Contestabile, A.; Ciani, E. Brain nitric oxide and its dual role in neurodegeneration/neuroprotection: Understanding molecular mechanisms to devise drug approaches. Curr. Med. Chem. 2003, 10, 2147–2174. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, C.J.; Padalko, E. iNOS (NOS2) at a glance. J. Cell Sci. 2004, 117, 2865–2867. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid. Redox Signal. 2011, 15, 2335–2381. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Mierla, A.L.; Minelli, A. Nrf2 and NF-κB and their concerted modulation in cancer pathogenesis and progression. Cancers 2010, 2, 483–497. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Kwon, O.K.; Kim, J.H.; Oh, S.R.; Kim, J.H.; Paik, J.H.; Marwoto, B.; Widjhati, R.; Juniarti, F.; Irawan, D.; et al. Rhododendron album Blume inhibits iNOS and COX-2 expression in LPS-stimulated RAW264.7 cells through the downregulation of NF-κB signalling. Int. J. Mol. Med. 2015, 35, 987–994. [Google Scholar] [PubMed]
- Zandi, E.; Karin, M. Bridging the gap: Composition, regulation, and physiological function of the IκB kinase complex. Mol. Cell. Biol. 1999, 19, 4547–4551. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Yamamoto, Y.; Wang, Q.M. The IKK NF-κB system: A treasure trove for drug development. Nat. Rev. Drug Discov. 2004, 3, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Leszek, J.; Barreto, G.E.; Gąsiorowski, K.; Koutsouraki, E.; Ávila-Rodrigues, M.; Aliev, G. Inflammatory mechanisms and oxidative stress as key factors responsible for progression of neurodegeneration: Role of brain innate immune system. CNS Neurol. Disord. Drug Targets 2016, 15, 1–8. [Google Scholar] [CrossRef]
- Gupta, S.P.; Yadav, S.; Singhal, N.K.; Tiwari, M.N.; Mishra, S.K.; Singh, M.P. Does restraining nitric oxide biosynthesis rescue from toxins-induced parkinsonism and sporadic Parkinson’s disease? Mol. Neurobiol. 2014, 49, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.P.; Donnelly, P.S.; Turner, B.J.; Hung, L.W.; Crouch, P.J.; Sherratt, N.A.; Tan, J.L.; Lim, N.K.; Lam, L.; Bica, L.; et al. Diacetylbis(N(4)-methylthiosemicarbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J. Biol. Chem. 2011, 286, 44035–44044. [Google Scholar] [CrossRef] [PubMed]
- Tsang, A.H.; Chung, K.K. Oxidative and nitrosative stress in Parkinson’s disease. Biochim. Biophys. Acta 2009, 1792, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Johnson, J.A. Nrf2—A therapeutic target for the treatment of neurodegenerative diseases. Free Radic. Biol. Med. 2015, 88, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar]
- Zipper, L.M.; Mulcahy, R.T. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J. Biol. Chem. 2002, 277, 36544–36552. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Keap1-Dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 2003, 278, 21592–21600. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-Activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Qu, J.; Shen, X. NF-κB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Grottelli, S.; Mierla, A.L.; Cacciatore, I.; Fornasari, E.; Roscini, L.; Cardinali, G.; Minelli, A. Neuroinflammation and endoplasmic reticulum stress are coregulated by cyclo(His-Pro) to prevent LPS neurotoxicity. Int. J. Biochem. Cell Biol. 2014, 51, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Bellezza, I.; Conte, C.; Culig, Z. Oxidative stress-related aging: A role for prostate cancer? Biochim. Biophys. Acta 2009, 1795, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bellezza, I.; Tucci, A.; Galli, F.; Grottelli, S.; Mierla, A.L.; Pilolli, F.; Minelli, A. Inhibition of NF-κB nuclear translocation via HO-1 activation underlies α-tocopheryl succinate toxicity. J. Nutr. Biochem. 2012, 23, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Kaur, U.; Banerjee, P.; Bir, A.; Sinha, M.; Biswas, A.; Chakrabarti, S. Reactive oxygen species, redox signaling and neuroinflammation in Alzheimer’s disease: The NF-κB connection. Curr. Top. Med. Chem. 2015, 15, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Turillazzi, E.; Neri, M.; Cerretani, D.; Cantatore, S.; Frati, P.; Moltoni, L.; Busardò, F.P.; Pomara, C.; Riezzo, I.; Fineschi, V. Lipid peroxidation and apoptotic response in rat brain areas induced by long-term administration of nandrolone: The mutual crosstalk between ROS and NF-kB. J. Cell. Mol. Med. 2016, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Piantadosi, C.A. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic. Biol. Med. 2008, 45, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Cha, Y.N.; Surh, Y.J. Peroxynitrite induces HO-1 expression via PI3K/Akt-dependent activation of NF-E2-related factor 2 in PC12 cells. Free Radic. Biol. Med. 2006, 41, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jang, J.; Na, H.; Cha, Y.; Surh, Y. Carbon monoxide produced by heme oxygenase-1 in response to nitrosative stress induces expression of glutamate-cysteine ligase in PC12 cells via activation of phosphatidylinositol 3-kinase and Nrf2 signaling. J. Biol. Chem. 2007, 282, 28577–28586. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-Related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Grottelli, S.; Bellezza, I.; Morozzi, G.; Peirce, M.J.; Marchetti, C.; Cacciatore, I.; Costanzi, E.; Minelli, A. Cyclo(His-Pro) Protects SOD1G93A Microglial Cells from Paraquat–Induced Toxicity. J. Clin. Cell. Immunol. 2015, 6, 1. [Google Scholar]
- Bernales, S.; Papa, F.R.; Walter, P. Intracellular signaling by the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2006, 22, 487–508. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Sovolyova, N.; Healy, S.; Samali, A.; Logue, S.E. Stressed to death–mechanisms of ER stress-induced cell death. Biol. Chem. 2014, 395, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Halliday, M.; Mallucci, G.R. Targeting the unfolded protein response in neurodegeneration: A new approach to therapy. Neuropharmacology 2014, 76, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Cozzolino, M.; Carrì, M.T. Old vs. new mechanisms in the pathogenesis of ALS. Brain Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kaus, A.; Sareen, D. ALS patient stem cells for unveiling disease signatures of motoneuron susceptibility: Perspectives on the deadly mitochondria, ER stress and calcium triad. Front. Cell. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef] [PubMed]
- Prentice, H.; Modi, J.P.; Wu, J.Y. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid. Med. Cell. Longev. 2015, 2015, 964518. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef] [PubMed]
- Karademir, B.; Corek, C.; Ozer, N.K. Endoplasmic reticulum stress and proteasomal system in amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2015, 88, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef] [PubMed]
- Ho, H.J.; Huang, D.Y.; Ho, F.M.; Lee, L.T.; Lin, W.W. Inhibition of lipopolysaccharide-induced inducible nitric oxide synthase expression by endoplasmic reticulum stress. Cell Signal. 2012, 24, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, Y.; Endo, M.; Tsukano, H.; Mori, M.; Oike, Y.; Gotoh, T. Molecular mechanisms of the LPS-induced non-apoptotic ER stress-CHOP pathway. J. Biochem. 2010, 147, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Hara, M.R.; Snyder, S.H. Cell signaling and neuronal death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 117–141. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Roy, N.; Liu, A.; Rajprohat, S.; Arnold, B.; Dukes, A.A.; Holbein, C.D.; Berman, S.B. Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and, in the presence of N-acetyl cysteine, mitophagy. Neurobiol. Dis. 2015, 74, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-what is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Sepers, M.D.; Raymond, L.A. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 2014, 19, 990–996. [Google Scholar] [CrossRef] [PubMed]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or overreaction? Exp. Neurol. 2016, 275, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.I.; Brennan-Minnella, A.M.; Won, S.J.; Shen, Y.; Hefner, C.; Shy, Y.; Sun, D.; Swanson, R.A. Intracellular pH reduction prevents excitotoxic and ischemic neuronal death by inhibiting NADPH oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, E4362–E4368. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Duchen, M.R. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta 2008, 1777, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Lau, A.; Tymianski, M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflügers Arch. 2010, 460, 525–542. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hanamsagar, R.; Bilbo, S.D. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J. Steroid Biochem. Mol. Biol. 2016, 160, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Yuste, J.E.; Tarragon, E.; Campuzano, C.M.; Ros-Bernal, F. Implications of glial nitric oxide in neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 322. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Kumar, A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp. Neurol. 2016, 275, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, G.; van Heijningen, S.; Reijne, A.C.; Nyakas, C.; van der Zee, E.A.; Eisel, U.L. Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front. Neurosci. 2015, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Minelli, A.; Grottelli, S.; Mierla, A.; Pinnen, F.; Cacciatore, I.; Bellezza, I. Cyclo(His-Pro) exerts anti-inflammatory effects by modulating NF-κB and Nrf2 signalling. Int. J. Biochem. Cell Biol. 2012, 44, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.; Brown, G.C. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: A dual-key mechanism of inflammatory neurodegeneration. J. Neuroinflamm. 2005, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 4, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.M.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
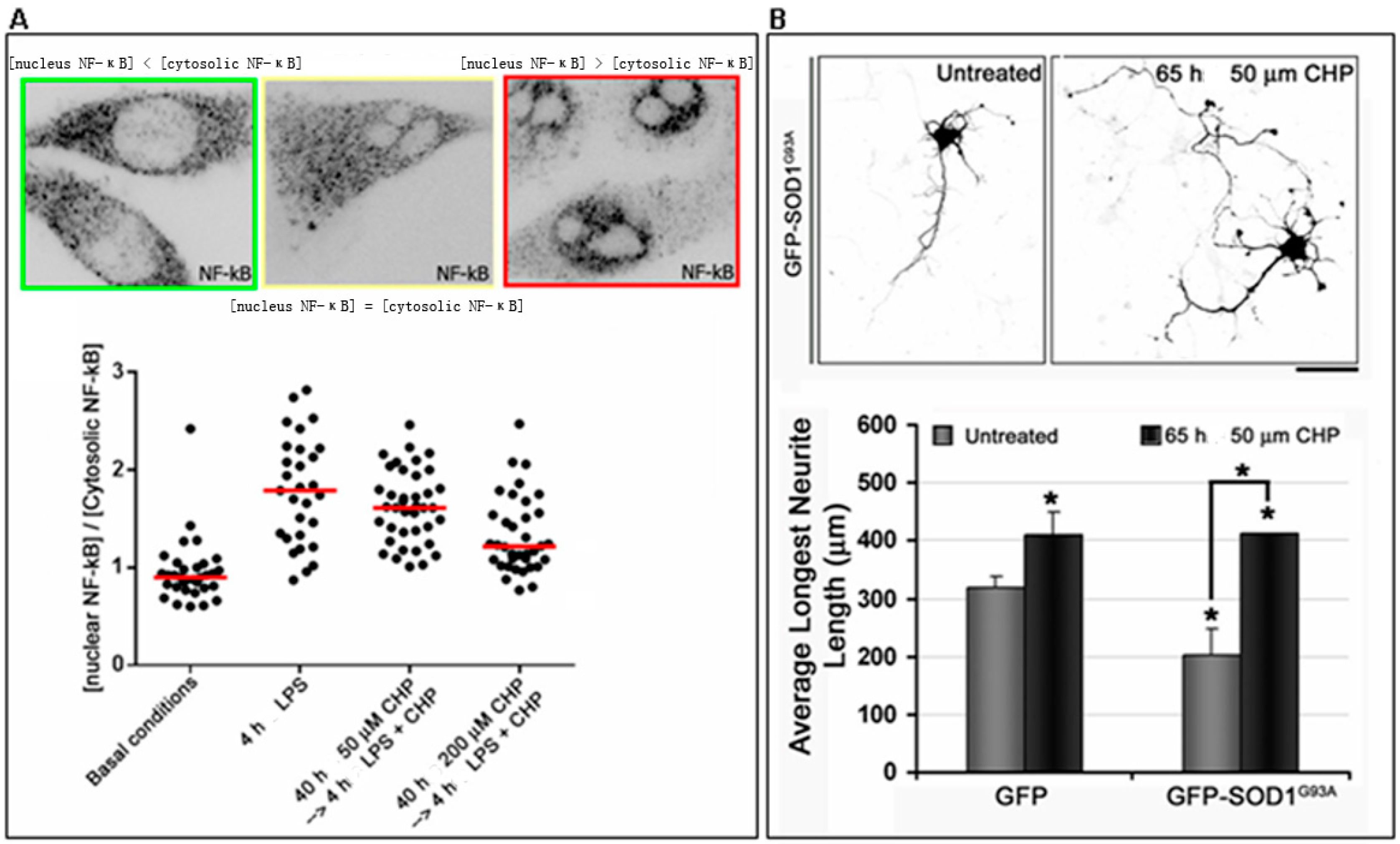
- Ferrari, I.; Minelli, A.; Pietrini, G. Role of cyclo(His-Pro) in familial amyotrophic lateral sclerosis (fALS) in vitro cell models. 2016. Unpublished data. [Google Scholar]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grottelli, S.; Ferrari, I.; Pietrini, G.; Peirce, M.J.; Minelli, A.; Bellezza, I. The Role of Cyclo(His-Pro) in Neurodegeneration. Int. J. Mol. Sci. 2016, 17, 1332. https://doi.org/10.3390/ijms17081332
Grottelli S, Ferrari I, Pietrini G, Peirce MJ, Minelli A, Bellezza I. The Role of Cyclo(His-Pro) in Neurodegeneration. International Journal of Molecular Sciences. 2016; 17(8):1332. https://doi.org/10.3390/ijms17081332
Chicago/Turabian StyleGrottelli, Silvia, Ilaria Ferrari, Grazia Pietrini, Matthew J. Peirce, Alba Minelli, and Ilaria Bellezza. 2016. "The Role of Cyclo(His-Pro) in Neurodegeneration" International Journal of Molecular Sciences 17, no. 8: 1332. https://doi.org/10.3390/ijms17081332
APA StyleGrottelli, S., Ferrari, I., Pietrini, G., Peirce, M. J., Minelli, A., & Bellezza, I. (2016). The Role of Cyclo(His-Pro) in Neurodegeneration. International Journal of Molecular Sciences, 17(8), 1332. https://doi.org/10.3390/ijms17081332