
Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD)

Abstract
:
1. Introduction
2. Results
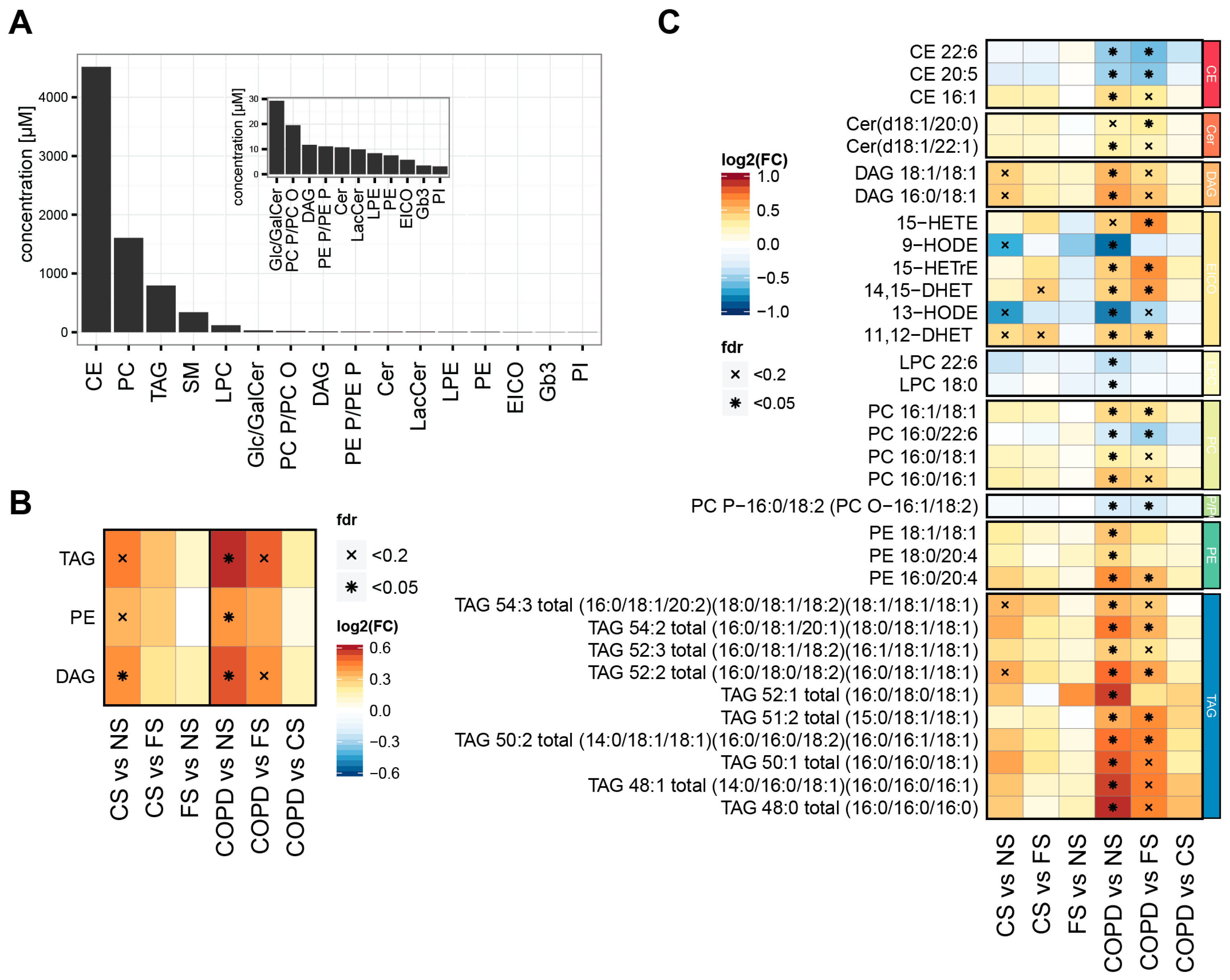
2.1. Smoking Status and Mild to Moderate COPD Are Reflected in the Serum Lipidome
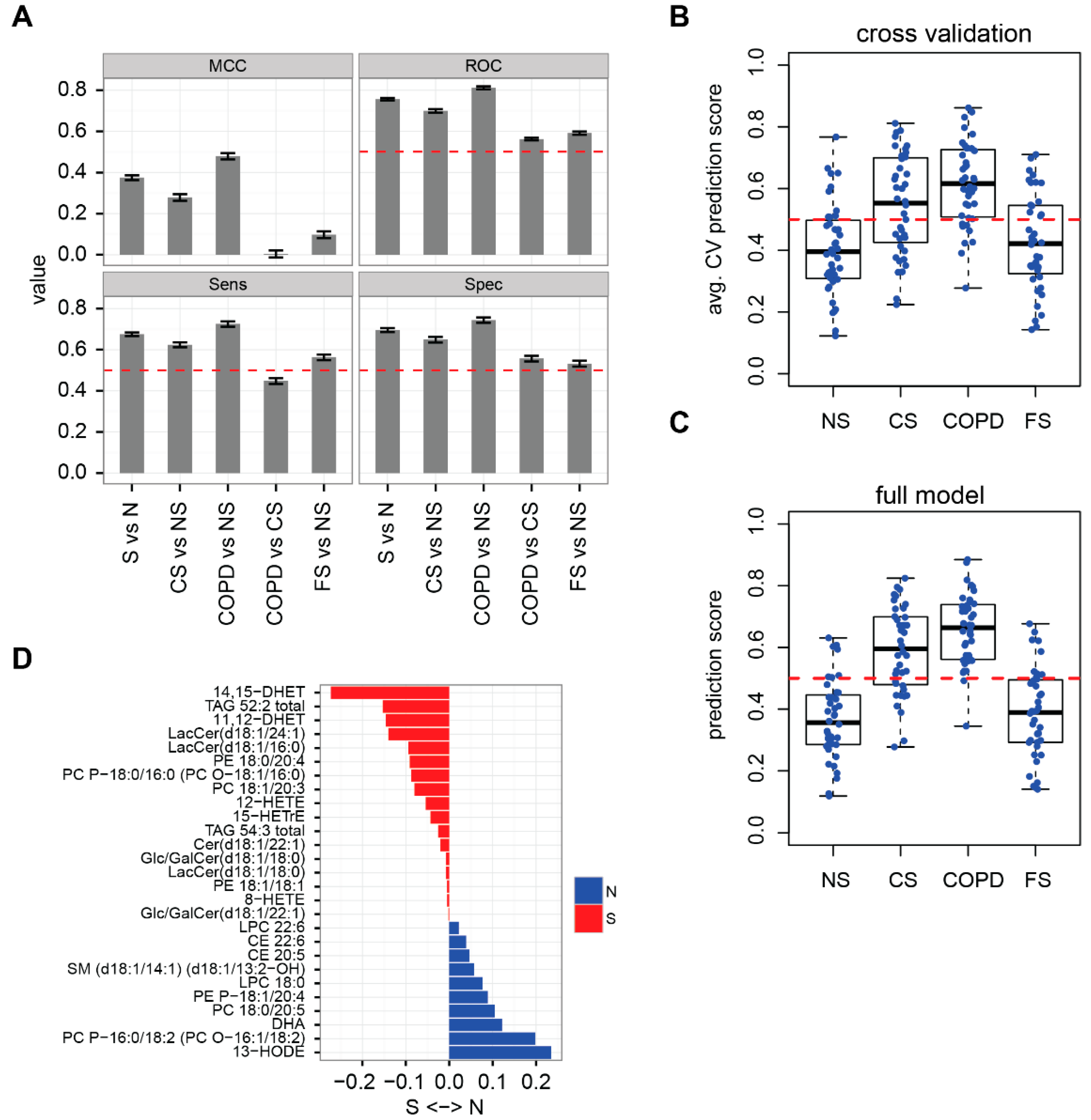
2.2. Lipidome Allows for Classification of Disease State and Smoking Status
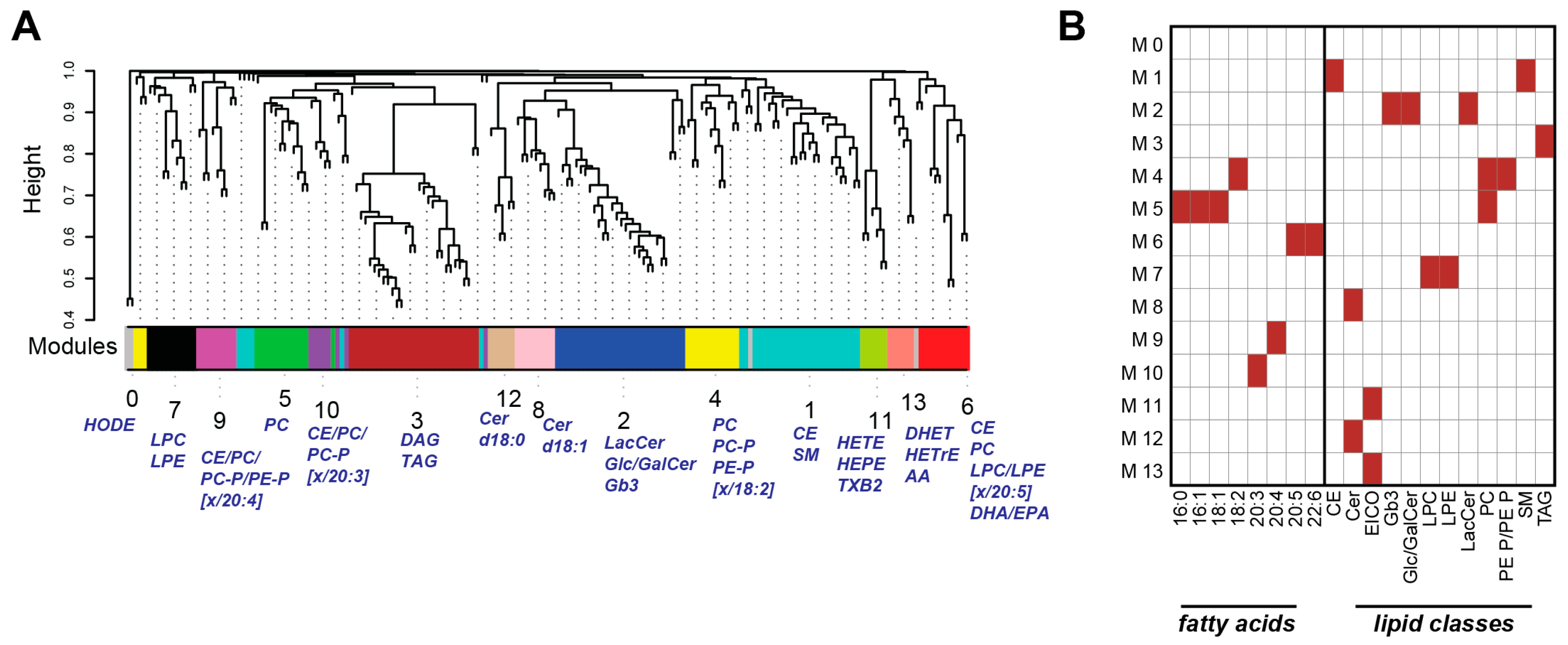
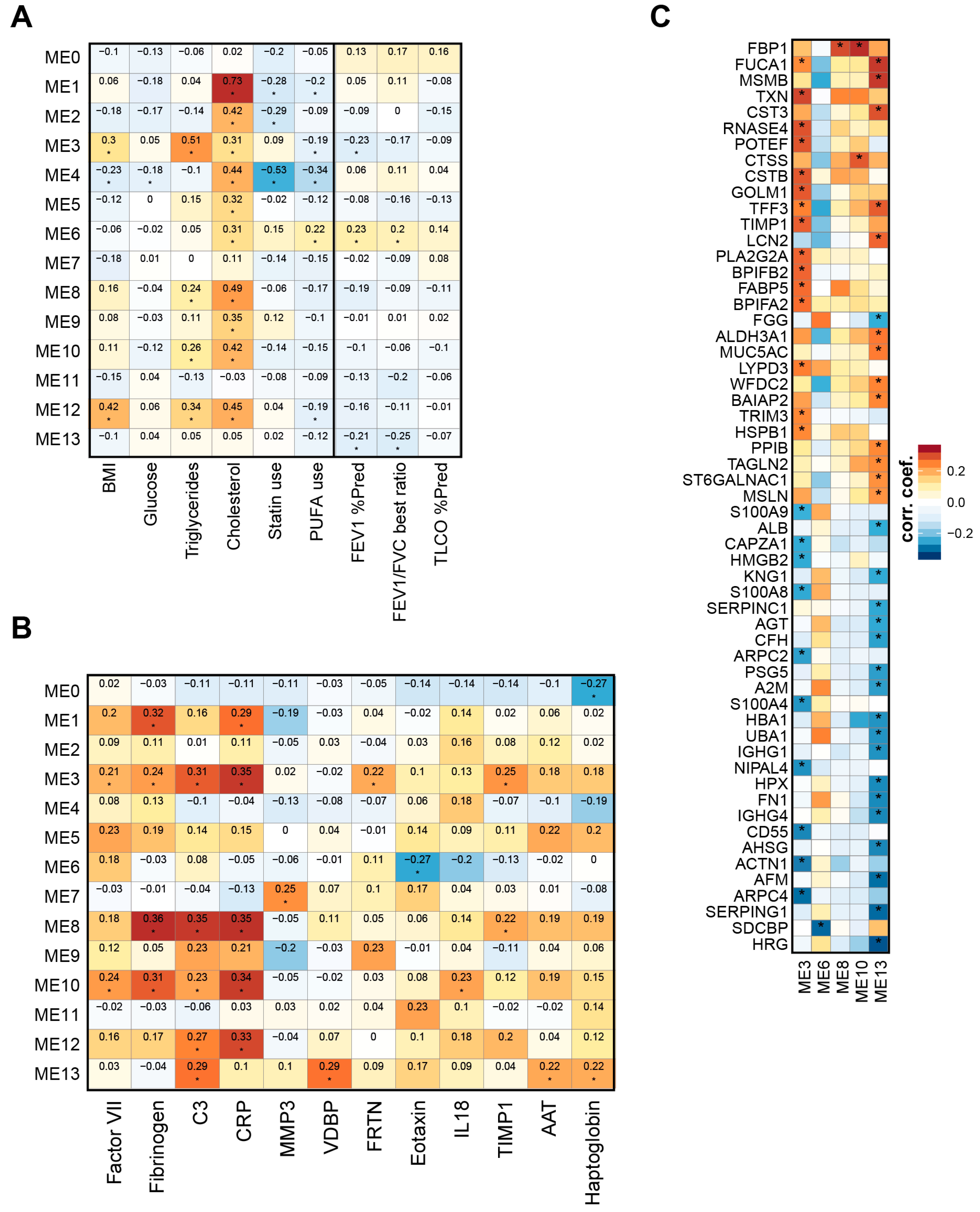
2.3. Weighted Correlation Network Analysis Reveals Specific Lipid Modules
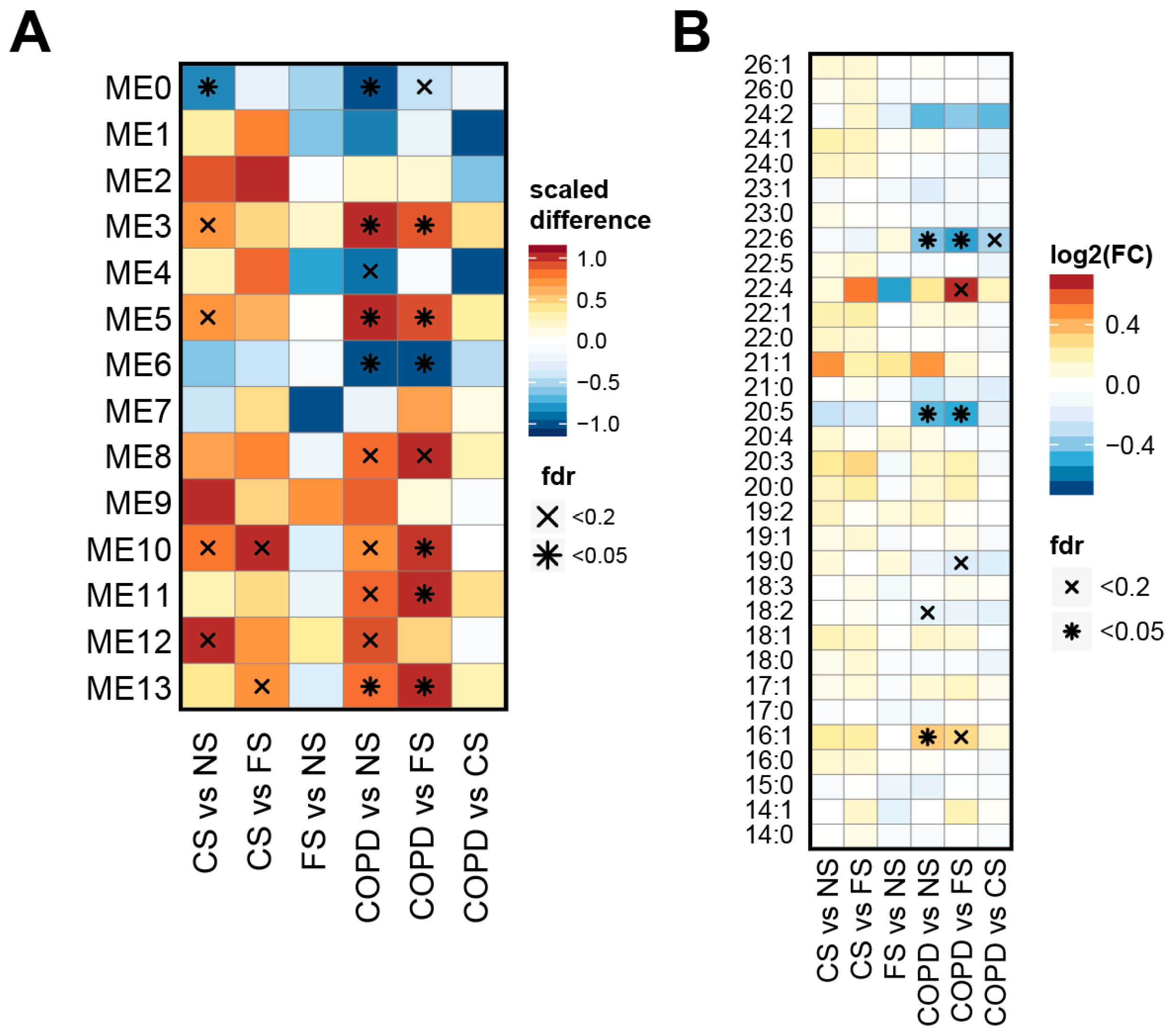
2.4. Lipid Module Profiles Reveal Group Differences
2.5. Lipid Modules Are Associated with Clinical Parameters and the Lung Response to Cigarette Smoking
3. Discussion
3.1. Previously Reported Effects
3.2. Lipidome Alterations in Our Study
3.3. Three Main Trends in Our Study
3.4. Effects on TAGs, DAGs, and PEs
3.5. Effects on PUFAs
3.6. Effects on Eicosanoids
3.7. Clinical Correlations
3.8. Strengths and Limitations
4. Materials and Methods
4.1. Subjects
4.2. Blood Collection and Lipidomics Analysis
4.3. Measurement of Plasma Markers
4.4. Computational Analyses of Lipidomics Data
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| CE | cholesteryl esters |
| Cer | ceramide |
| CS | current smoker group |
| DAG | diacylglycerol (fatty acyl groups separated with a hyphen, e.g., PC 16:0-18:1) |
| DHA | docosahexaenoic acid |
| DHET | dihydroxyeicosatrienoic acid |
| EPA | eicosapentaenoic acid |
| FA | fatty acid |
| FS | former smoker group |
| Gb3 | globotriaosylceramide |
| GlcCer | glucosylceramide |
| HETE | hydroxyeicosatetraenoic acid |
| HODE | hydroxyoctadecadienoic acid |
| LacCer | lactosylceramide |
| LPC/LPE | lyso PC/PE |
| MUFA | monounsaturated fatty acid |
| NS | never-smoker group |
| PA | phosphatidic acid |
| PC O/PE O | ether-linked alkyl phospholipid |
| PC P/PE P | ether-linked alkenyl phospholipid |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PG | phosphatidylglycerol |
| PI | phosphatidylinositol |
| PS | phosphatidylserine |
| PUFA | polyunsaturated fatty acid |
| SM | sphingomyelin |
| TAG | triacylglycerol |
References
- Vestbo, J.; Hurd, S.S.; Agusti, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Guzman, E.; Mannino, D.M. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin. Chest Med. 2014, 35, 7–16. [Google Scholar] [CrossRef] [PubMed]
- WHO. The 10 Leading Causes of Death in the World, 2000 and 2012. Available online: http://www.who.int/mediacentre/factsheets/fs310/en/ (accessed on 15 September 2016).
- Gold, D.R.; Wang, X.; Wypij, D.; Speizer, F.E.; Ware, J.H.; Dockery, D.W. Effects of cigarette smoking on lung function in adolescent boys and girls. N. Engl. J. Med. 1996, 335, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Langhammer, A.; Johnsen, R.; Gulsvik, A.; Holmen, T.L.; Bjermer, L. Sex differences in lung vulnerability to tobacco smoking. Eur. Respir. J. 2003, 21, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Hooper, R.; Burney, P.; Vollmer, W.M.; McBurnie, M.A.; Gislason, T.; Tan, W.C.; Jithoo, A.; Kocabas, A.; Welte, T.; Buist, A.S. Risk factors for COPD spirometrically defined from the lower limit of normal in the BOLD project. Eur. Respir. J. 2012, 39, 1343–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vestbo, J.; Agusti, A.; Wouters, E.F.; Bakke, P.; Calverley, P.M.; Celli, B.; Coxson, H.; Crim, C.; Edwards, L.D.; Locantore, N.; et al. Should we view chronic obstructive pulmonary disease differently after ECLIPSE? A clinical perspective from the study team. Am. J. Respir. Crit. Care Med. 2014, 189, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Sewer, A.; Schneider, T.; Elamin, A.; Martin, F.; Dijon, S.; Luettich, K.; Guedj, E.; Vuillaume, G.; Ivanov, N.V. Alterations in the sputum proteome and transcriptome in smokers and early-stage COPD subjects. J. Proteom. 2015, 128, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Ockene, I.S.; Miller, N.H. Cigarette smoking, cardiovascular disease, and stroke a statement for healthcare professionals from the American Heart Association. Circulation 1997, 96, 3243–3247. [Google Scholar] [CrossRef] [PubMed]
- Pujades-Rodriguez, M.; George, J.; Shah, A.D.; Rapsomaniki, E.; Denaxas, S.; West, R.; Smeeth, L.; Timmis, A.; Hemingway, H. Heterogeneous associations between smoking and a wide range of initial presentations of cardiovascular disease in 1,937,360 people in England: Lifetime risks and implications for risk prediction. Int. J. Epidemiol. 2015, 44, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Maclay, J.D.; MacNee, W. Cardiovascular disease in COPD: Mechanisms. CHEST J. 2013, 143, 798–807. [Google Scholar] [CrossRef] [PubMed]
- De Leon, H.; Boue, S.; Szostak, J.; Peitsch, M.C.; Hoeng, J. Systems biology research into cardiovascular Disease: Contributions of lipidomics-based approaches to biomarker discovery. Curr. Drug Discov. Technol. 2015, 12, 129–154. [Google Scholar] [CrossRef] [PubMed]
- Messner, B.; Bernhard, D. Smoking and cardiovascular disease mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Shahar, E.; Boland, L.L.; Folsom, A.R.; Tockman, M.S.; McGovern, P.G.; Eckfeldt, J.H. Docosahexaenoic acid and smoking-related chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1999, 159, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Shahar, E.; Folsom, A.R.; Melnick, S.L.; Tockman, M.S.; Comstock, G.W.; Gennaro, V.; Higgins, M.W.; Sorlie, P.D.; Ko, W.J.; Szklo, M. Dietary n-3 polyunsaturated fatty acids and smoking-related chronic obstructive pulmonary disease. N. Engl. J. Med. 1994, 331, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.A.; Fong, J.; Bemert, J.T.; Browner, W.S. Relation of smoking and alcohol consumption to serum fatty acids. Am. J. Epidemiol. 1996, 144, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Fernando, H.; Bhopale, K.K.; Kondraganti, S.; Kaphalia, B.S.; Shakeel Ansari, G.A. Lipidomic changes in rat liver after long-term exposure to ethanol. Toxicol. Appl. Pharmacol. 2011, 255, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.; Gonzalez, L.; Catala, A. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog. Lipid Res. 2005, 44, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Blanksby, S.J.; Mitchell, T.W. Advances in mass spectrometry for lipidomics. Annu. Rev. Anal. Chem. 2010, 3, 433–465. [Google Scholar] [CrossRef] [PubMed]
- Wang-Sattler, R.; Yu, Y.; Mittelstrass, K.; Lattka, E.; Altmaier, E.; Gieger, C.; Ladwig, K.H.; Dahmen, N.; Weinberger, K.M.; Hao, P. Metabolic profiling reveals distinct variations linked to nicotine consumption in humans—First results from the KORA study. PLoS ONE 2008, 3, e3863. [Google Scholar] [CrossRef] [PubMed]
- Weir, J.M.; Wong, G.; Barlow, C.K.; Greeve, M.A.; Kowalczyk, A.; Almasy, L.; Comuzzie, A.G.; Mahaney, M.C.; Jowett, J.B.; Shaw, J.; et al. Plasma lipid profiling in a large population-based cohort. J. Lipid Res. 2013, 54, 2898–2908. [Google Scholar] [CrossRef] [PubMed]
- Müller, D.C.; Degen, C.; Scherer, G.; Jahreis, G.; Niessner, R.; Scherer, M. Metabolomics using GC–TOF–MS followed by subsequent GC–FID and HILIC–MS/MS analysis revealed significantly altered fatty acid and phospholipid species profiles in plasma of smokers. J. Chromatogr. B 2014, 966, 117–126. [Google Scholar]
- Ubhi, B.K.; Riley, J.H.; Shaw, P.A.; Lomas, D.A.; Tal-Singer, R.; MacNee, W.; Griffin, J.L.; Connor, S.C. Metabolic profiling detects biomarkers of protein degradation in COPD patients. Eur. Respir. J. 2012, 40, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.; Talikka, M.; Hoeng, J.; Peitsch, M. Identification of gene expression signature for cigarette smoke exposure response—From man to mouse. Hum. Exp. Toxicol. 2015, 34, 1200–1211. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.I.; Luettich, K.; Peck, M.J.; Pierri, E.; Felber-Medlin, L.; Vuillaume, G.; Leroy, P.; Peitsch, M.C. Physiological measures in subjects with mild to moderate COPD compared to asymptomatic current smokers, former smokers and never-smokers. Unpublished work. 2016. [Google Scholar]
- Talikka, M.; Martin, F.; Sewer, A; Vuillaume, G.; Leroy, P.; Chaudhary, N.; Peck, M.J.; Peitsch, M.C.; Hoeng, J. Systems toxicology approach to evaluate the impact of smoking and chronic obstructive pulmonary disease on the nasal epithelium. 2016; submitted. [Google Scholar]
- Quehenberger, O.; Armando, A.M.; Brown, A.H.; Milne, S.B.; Myers, D.S.; Merrill, A.H.; Bandyopadhyay, S.; Jones, K.N.; Kelly, S.; Shaner, R.L. Lipidomics reveals a remarkable diversity of lipids in human plasma. J. Lipid Res. 2010, 51, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Gray, K.A.; Seal, R.L.; Tweedie, S.; Wright, M.W.; Bruford, E.A. A review of the new HGNC gene family resource. Hum. Genom. 2016, 10, 6. [Google Scholar] [CrossRef] [PubMed]
- Samols, D.; Agrawal, A.; Kushner, I. Acute phase proteins. In Cytokine Reference on-Line; Oppenheim, J.J., Feldman, M., Eds.; Academic Press: London, UK, 2002. [Google Scholar]
- Cantin, A.M. Cellular response to cigarette smoke and oxidants: Adapting to survive. Proc. Am. Thorac. Soc. 2010, 7, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, S.L.; Balgoma, D.; Wheelock, A.M.; Haeggstrom, J.Z.; Dahlen, S.E.; Wheelock, C.E. Lipid mediator profiling in pulmonary disease. Curr. Pharm. Biotechnol. 2011, 12, 1026–1052. [Google Scholar] [CrossRef]
- Telenga, E.D.; Hoffmann, R.F.; Ruben, T.K.; Hoonhorst, S.J.; Willemse, B.W.; van Oosterhout, A.J.; Heijink, I.H.; van den Berge, M.; Jorge, L.; Sandra, P.; et al. Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. Am. J. Respir. Crit. Care Med. 2014, 190, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Titz, B.; Boue, S.; Phillips, B.; Talikka, M.; Vihervaara, T.; Schneider, T.; Nury, C.; Elamin, A.; Guedj, E.; Peck, M.J.; et al. Effects of cigarette smoke, cessation and switching to two heat-not-burn tobacco products on lung lipid metabolism in C57BL/6 and Apoe−/− mice—An integrative systems toxicology analysis. Toxicol. Sci. Off. J. Soc. Toxicol. 2016, 149, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Holzapfel, C.; Dong, X.; Bader, E.; Yu, Z.; Prehn, C.; Perstorfer, K.; Jaremek, M.; Roemisch-Margl, W.; Rathmann, W.; et al. Effects of smoking and smoking cessation on human serum metabolite profile: Results from the KORA cohort study. BMC Med. 2013, 11, 60. [Google Scholar] [CrossRef] [PubMed]
- Vestbo, J.; Anderson, W.; Coxson, H.O.; Crim, C.; Dawber, F.; Edwards, L.; Hagan, G.; Knobil, K.; Lomas, D.A.; MacNee, W.; et al. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-points (ECLIPSE). Eur. Respir. J. 2008, 31, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Deeb, R.S.; Ma, Y.; Staudt, M.R.; Crystal, R.G.; Gross, S.S. Serum metabolite biomarkers discriminate healthy smokers from COPD smokers. PLoS ONE 2015, 10, e0143937. [Google Scholar] [CrossRef] [PubMed]
- Craig, W.Y.; Palomaki, G.E.; Haddow, J.E. Cigarette smoking and serum lipid and lipoprotein concentrations: An analysis of published data. BMJ 1989, 298, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Campbell, S.C.; Moffatt, R.J.; Stamford, B.A. Smoking and smoking cessation—The relationship between cardiovascular disease and lipoprotein metabolism: A review. Atherosclerosis 2008, 201, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. ω-3 Polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology? Br. J. Clin. Pharmacol. 2013, 75, 645–662. [Google Scholar] [CrossRef] [PubMed]
- Fowkes, F.; Housley, E.; Cawood, E.; Macintyre, C.; Ruckley, C.; Prescott, R. Edinburgh artery study: Prevalence of asymptomatic and symptomatic peripheral arterial disease in the general population. Int. J. Epidemiol. 1991, 20, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Leng, G.; Horrobin, D.; Fowkes, F.; Smith, F.; Lowe, G.; Donnan, P.; Ells, K. Plasma essential fatty acids, cigarette smoking, and dietary antioxidants in peripheral arterial disease. A population-based case-control study. Arterioscler. Thromb. Vasc. Biol. 1994, 14, 471–478. [Google Scholar] [CrossRef]
- Scaglia, N.; Chatkin, J.; Chapman, K.R.; Ferreira, I.; Wagner, M.; Selby, P.; Allard, J.; Zamel, N. The relationship between ω-3 and smoking habit: A cross-sectional study. Lipids Health Dis. 2016, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Wood, L.G. ω-3 Polyunsaturated fatty acids and chronic obstructive pulmonary disease. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Chambless, L.E.; Heiss, G.; Folsom, A.R.; Rosamond, W.; Szklo, M.; Sharrett, A.R.; Clegg, L.X. Association of coronary heart disease incidence with carotid arterial wall thickness and major risk factors: The Atherosclerosis Risk in Communities (ARIC) Study, 1987–1993. Am. J. Epidemiol. 1997, 146, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.S.; Rothwell, S.T.; Madans, J.H.; Finucane, F.F.; Freid, V.; Kleinman, J.C.; Barbano, H.E.; Feldman, J.J. Plan and operation of the NHANES I Epidemiologic Followup Study, 1987. Vital Health Stat. 1 1992, 27, 1–190. [Google Scholar] [PubMed]
- Schwartz, J. Role of polyunsaturated fatty acids in lung disease. Am. J. Clin. Nutr. 2000, 71, 393S–396S. [Google Scholar]
- Hirayama, F.; Lee, A.H.; Binns, C.W.; Hiramatsu, N.; Mori, M.; Nishimura, K. Dietary intake of isoflavones and polyunsaturated fatty acids associated with lung function, breathlessness and the prevalence of chronic obstructive pulmonary disease: Possible protective effect of traditional Japanese diet. Mol. Nutr. Food Res. 2010, 54, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.P.; Niti, M.; Yap, K.B.; Tan, W.C. Dietary and supplemental antioxidant and anti-inflammatory nutrient intakes and pulmonary function. Public Health Nutr. 2014, 17, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Fulton, A.S.; Hill, A.M.; Williams, M.T.; Howe, P.R.; Coates, A.M. Paucity of evidence for a relationship between long-chain ω-3 fatty acid intake and chronic obstructive pulmonary disease: A systematic review. Nutr. Rev. 2015, 73, 612–623. [Google Scholar] [CrossRef] [PubMed]
- Atlantis, E.; Cochrane, B. The association of dietary intake and supplementation of specific polyunsaturated fatty acids with inflammation and functional capacity in chronic obstructive pulmonary disease: A systematic review. Int. J. Evid. Based Healthc. 2016, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Wallner, S.; Schmitz, G. Plasmalogens the neglected regulatory and scavenging lipid species. Chem. Phys. Lipids 2011, 164, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Spiteller, G. Peroxyl radicals: Inductors of neurodegenerative and other inflammatory diseases. Their origin and how they transform cholesterol, phospholipids, plasmalogens, polyunsaturated fatty acids, sugars, and proteins into deleterious products. Free Radic. Biol. Med. 2006, 41, 362–387. [Google Scholar] [CrossRef] [PubMed]
- De Castro, J.; Hernández-Hernández, A.; Rodríguez, M.C.; Sardina, J.L.; Llanillo, M.; Sánchez-Yagüe, J. Comparison of changes in erythrocyte and platelet phospholipid and fatty acid composition and protein oxidation in chronic obstructive pulmonary disease and asthma. Platelets 2007, 18, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-R.; Chida, A.S.; Bauter, M.R.; Shafiq, N.; Seweryniak, K.; Maggirwar, S.B.; Kilty, I.; Rahman, I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and posttranslational modifications of histone deacetylase in macrophages. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2006, 291, L46–L57. [Google Scholar] [CrossRef] [PubMed]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stampfli, M.R. Disruption of pulmonary lipid homeostasis drives cigarette smoke-induced lung inflammation in mice. Eur. Respir. J. 2015, 46, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Yanbaeva, D.G.; Dentener, M.A.; Creutzberg, E.C.; Wesseling, G.; Wouters, E.F. Systemic effects of smoking. Chest J. 2007, 131, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Hallman, A.H.; Yousif, M.K.; Chien, M.T. Oxidative stress, respiratory muscle dysfunction, and potential therapeutics in chronic obstructive pulmonary disease. Front. Biol. 2012, 7, 506–513. [Google Scholar] [CrossRef]
- Powell, W.S.; Rokach, J. Biosynthesis, biological effects, and receptors of hydroxyeicosatetraenoic acids (HETEs) and oxoeicosatetraenoic acids (oxo-ETEs) derived from arachidonic acid. Biochim. Biophys. Acta 2015, 1851, 340–355. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, P.; Hamberg, M.; Diczfalusy, U. Formation of 15-HETE as a major hydroxyeicosatetraenoic acid in the atherosclerotic vessel wall. Biochim. Biophys. Acta 1985, 834, 272–274. [Google Scholar] [CrossRef]
- Kundumani-Sridharan, V.; Dyukova, E.; Hansen, D.E.; Rao, G.N. 12/15-Lipoxygenase mediates high-fat diet-induced endothelial tight junction disruption and monocyte transmigration: A new role for 15 (S)-hydroxyeicosatetraenoic acid in endothelial cell dysfunction. J. Biol. Chem. 2013, 288, 15830–15842. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.S.; Kim, M.S.; Lee, D.H.; Oh, D.K.; Yoon, D.Y. 15-Hydroxyeicosatetraenoic acid inhibits phorbol-12-myristate-13-acetate-induced MUC5AC expression in NCI-H292 respiratory epithelial cells. J. Microbiol. Biotechnol. 2015, 25, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A.; Norris, A.W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007, 292, C996–C1012. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Kaduce, T.L.; Weintraub, N.L.; Harmon, S.; Teesch, L.M.; Morisseau, C.; Thompson, D.A.; Hammock, B.D.; Spector, A.A. Pathways of epoxyeicosatrienoic acid metabolism in endothelial cells: Implications for the vascular effects of soluble epoxide hydrolase inhibition. J. Biol. Chem. 2001, 276, 14867–14874. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Katakam, P.V.G.; VanRollins, M.; Weintraub, N.L.; Spector, A.A.; Lee, H.-C. Dihydroxyeicosatrienoic acids are potent activators of Ca2+-activated K+ channels in isolated rat coronary arterial myocytes. J. Physiol. 2001, 534, 651–667. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Oguro, A.; Osada-Oka, M.; Funae, Y.; Imaoka, S. Epoxyeicosatrienoic acids and/or their metabolites promote hypoxic response of cells. J. Pharmacol. Sci. 2008, 108, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Hu, S.; Xu, B.; Snyder, G.D.; Harmon, S.; Yao, J.; Liu, Y.; Sangras, B.; Falck, J.R.; Weintraub, N.L.; et al. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-α. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H55–H63. [Google Scholar] [CrossRef] [PubMed]
- Podolin, P.L.; Bolognese, B.J.; Foley, J.F.; Long Iii, E.; Peck, B.; Umbrecht, S.; Zhang, X.; Zhu, P.; Schwartz, B.; Xie, W.; et al. In vitro and in vivo characterization of a novel soluble epoxide hydrolase inhibitor. Prostaglandins Other Lipid Mediat. 2013, 104–105, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.R.; Pinkerton, K.E.; Watanabe, T.; Pedersen, T.L.; Ma, S.J.; Hammock, B.D. Attenuation of tobacco smoke-induced lung inflammation by treatment with a soluble epoxide hydrolase inhibitor. Proc. Natl. Acad. Sci. USA 2005, 102, 2186–2191. [Google Scholar] [CrossRef] [PubMed]
- Dandona, P.; Mohanty, P.; Ghanim, H.; Aljada, A.; Browne, R.; Hamouda, W.; Prabhala, A.; Afzal, A.; Garg, R. The suppressive effect of dietary restriction and weight loss in the obese on the generation of reactive oxygen species by leukocytes, lipid peroxidation, and protein carbonylation. J. Clin. Endocrinol. Metab. 2001, 86, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Umeno, A.; Shichiri, M. Lipid peroxidation biomarkers for evaluating oxidative stress and assessing antioxidant capacity in vivo. J. Clin. Biochem. Nutr. 2013, 52, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Niki, E. Detection of lipid peroxidation in vivo: Total hydroxyoctadecadienoic acid and 7-hydroxycholesterol as oxidative stress marker. Free Radic. Res. 2004, 38, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Rise, P.; Ghezzi, S.; Galli, C. Relative potencies of statins in reducing cholesterol synthesis and enhancing linoleic acid metabolism. Eur. J. Pharmacol. 2003, 467, 73–75. [Google Scholar] [CrossRef]
- Jula, A.; Marniemi, J.; Ronnemaa, T.; Virtanen, A.; Huupponen, R. Effects of diet and simvastatin on fatty acid composition in hypercholesterolemic men: A randomized controlled trial. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1952–1959. [Google Scholar] [CrossRef] [PubMed]
- Nordestgaard, B.G.; Langsted, A.; Mora, S.; Kolovou, G.; Baum, H.; Bruckert, E.; Watts, G.F.; Sypniewska, G.; Wiklund, O.; Boren, J.; et al. Fasting is not routinely required for determination of a lipid profile: Clinical and laboratory implications including flagging at desirable concentration cut-points—A joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur. Heart J. 2016, 37, 1944–1958. [Google Scholar] [PubMed]
- Mora, S. Nonfasting for routine lipid testing: From evidence to action. JAMA Intern. Med. 2016, 176, 1005–1006. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, K. Unraveling Glycerophospholipidomes by Lipidomics. In Biomarker Methods in Drug Discovery and Development; Wang, F., Ed.; Humana Press: New York, NY, USA, 2008; pp. 369–384. [Google Scholar]
- Ståhlman, M.; Ejsing, C.S.; Tarasov, K.; Perman, J.; Borén, J.; Ekroos, K. High-throughput shotgun lipidomics by quadrupole time-of-flight mass spectrometry. J. Chromatogr. B 2009, 877, 2664–2672. [Google Scholar]
- Ejsing, C.S.; Duchoslav, E.; Sampaio, J.; Simons, K.; Bonner, R.; Thiele, C.; Ekroos, K.; Shevchenko, A. Automated Identification and quantification of glycerophospholipid molecular species by multiple precursor ion scanning. Anal. Chem. 2006, 78, 6202–6214. [Google Scholar] [CrossRef] [PubMed]
- Ekroos, K.; Chernushevich, I.V.; Simons, K.; Shevchenko, A. Quantitative profiling of phospholipids by multiple precursor ion scanning on a hybrid quadrupole time-of-flight mass spectrometer. Anal. Chem. 2002, 74, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Hermansson, M.; Uphoff, A.; Käkelä, R.; Somerharju, P. Automated quantitative analysis of complex lipidomes by liquid chromatography/mass spectrometry. Anal. Chem. 2005, 77, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Baumer, K.; Boue, S.; Dijon, S.; Dulize, R.; Ekroos, K.; Elamin, A.; Foong, C.; Guedj, E.; Hoeng, J.; et al. Comprehensive systems biology analysis of a 7-month cigarette smoke inhalation study in C57BL/6 mice. Sci. Data 2016, 3, 150077. [Google Scholar] [CrossRef] [PubMed]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Available online: https://cran.r-project.org/doc/manuals/r-release/fullrefman.pdf (accessed on 15 September 2016).
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, i05. [Google Scholar] [CrossRef]
- Powers, D.M. Evaluation: From precision, recall and F-measure to ROC, informedness, markedness and correlation. J. Mach. Learn. Technol. 2011, 2, 37–63. [Google Scholar]
- Väremo, L.; Nielsen, J.; Nookaew, I. Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 2013, 41, 4378–4391. [Google Scholar]
- Fahy, E.; Subramaniam, S.; Murphy, R.C.; Nishijima, M.; Raetz, C.R.; Shimizu, T.; Spener, F.; van Meer, G.; Wakelam, M.J.; Dennis, E.A. Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 2009, 50, S9–S14. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NS (Never Smoker) | CS (Current Smoker) | COPD | FS (Former Smoker) | |
|---|---|---|---|---|
| N (Filtered a/Total) | 39/40 | 39/40 | 39/40 | 38/40 |
| GOLD 1/GOLD 2 | 0/0 | 0/0 | 19/20 | 0/0 |
| Sex (Male/Female) | 21/18 | 22/17 | 22/17 | 21/17 |
| Age (Years) | 55.77 ± 6.81 | 55.51 ± 6.79 | 57.87 ± 6.91 | 57.08 ± 6.98 |
| BMI (Body Mass Index) (kg/m2) | 26.67 ± 4.13 | 27.38 ± 3.31 | 26.53 ± 3.96 | 27.22 ± 3.08 |
| Smoking History (Pack-Years) | 0 ± 0 | 34.21 ± 14.69 | 45.6 ± 22.7 | 27.72 ± 13.38 |
| Quitting Duration (Years) | 0 ± 0 | 0 ± 0 | 0 ± 0 | 13.97 ± 10.46 |
| FEV1 b (% Predicted) | 110.55 ± 11.51 | 102.07 ± 11.63 | 75.44 ± 18.18 | 108.52 ± 12.2 |
| FVC c (% Predicted) | 122.56 ± 14.75 | 118.78 ± 13.41 | 110.53 ± 18.84 | 122.3 ± 12.63 |
| FEV1/FVC (%) | 73.9 ± 5.83 | 70.21 ± 5.19 | 54.96 ± 7.93 | 72.03 ± 4.54 |
| TLCO d (% Predicted) | 94.2 ± 11.96 | 80.49 ± 13.08 | 67.51 ± 17.24 | 91.14 ± 13.81 |
| Lipid Modifier Drugs (Statins) | 5% | 5% | 28% | 24% |
| Platform | Monitored Lipid Classes |
|---|---|
| Shotgun lipidomics | Cholesterol esters Phosphatidylcholines Lysophosphatidylcholines and other lysophospholipids Ether-linked phosphatidylcholines and other ether-linked phospholipids Phosphatidylserines Phosphatidylethanolamines Phosphatidylglycerols Phosphatidylinositiols Phosphatidic acid Sphingomyelins Diacylglycerols |
| Triacylglycerol lipidomics | Triacylglycerols |
| Ceramide and cerebroside lipidomics | Ceramides Cerebrosides (Lactosylceramides; Galactosyl- and Glucosylceramides; Globotriaosylceramides) |
| Eicosanoid lipidomics | Arachidonic acid Eicosapentaenoic acid Docosahexaenoic acid Prostaglandins Thromboxanes Hydroxyeicosapentaenoic acids Hydroxyeicosatetraenoic acids Dihydroxyeicosatrienoic acids Hydroxyoctadecadienoic acids Hydroxyoctadecatrienoic acids Leukotrienes Lipoxines |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titz, B.; Luettich, K.; Leroy, P.; Boue, S.; Vuillaume, G.; Vihervaara, T.; Ekroos, K.; Martin, F.; Peitsch, M.C.; Hoeng, J. Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD). Int. J. Mol. Sci. 2016, 17, 1583. https://doi.org/10.3390/ijms17091583
Titz B, Luettich K, Leroy P, Boue S, Vuillaume G, Vihervaara T, Ekroos K, Martin F, Peitsch MC, Hoeng J. Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD). International Journal of Molecular Sciences. 2016; 17(9):1583. https://doi.org/10.3390/ijms17091583
Chicago/Turabian StyleTitz, Bjoern, Karsta Luettich, Patrice Leroy, Stephanie Boue, Gregory Vuillaume, Terhi Vihervaara, Kim Ekroos, Florian Martin, Manuel C. Peitsch, and Julia Hoeng. 2016. "Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD)" International Journal of Molecular Sciences 17, no. 9: 1583. https://doi.org/10.3390/ijms17091583
APA StyleTitz, B., Luettich, K., Leroy, P., Boue, S., Vuillaume, G., Vihervaara, T., Ekroos, K., Martin, F., Peitsch, M. C., & Hoeng, J. (2016). Alterations in Serum Polyunsaturated Fatty Acids and Eicosanoids in Patients with Mild to Moderate Chronic Obstructive Pulmonary Disease (COPD). International Journal of Molecular Sciences, 17(9), 1583. https://doi.org/10.3390/ijms17091583