
Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis

{kind=link}
Abstract
:1. Introduction
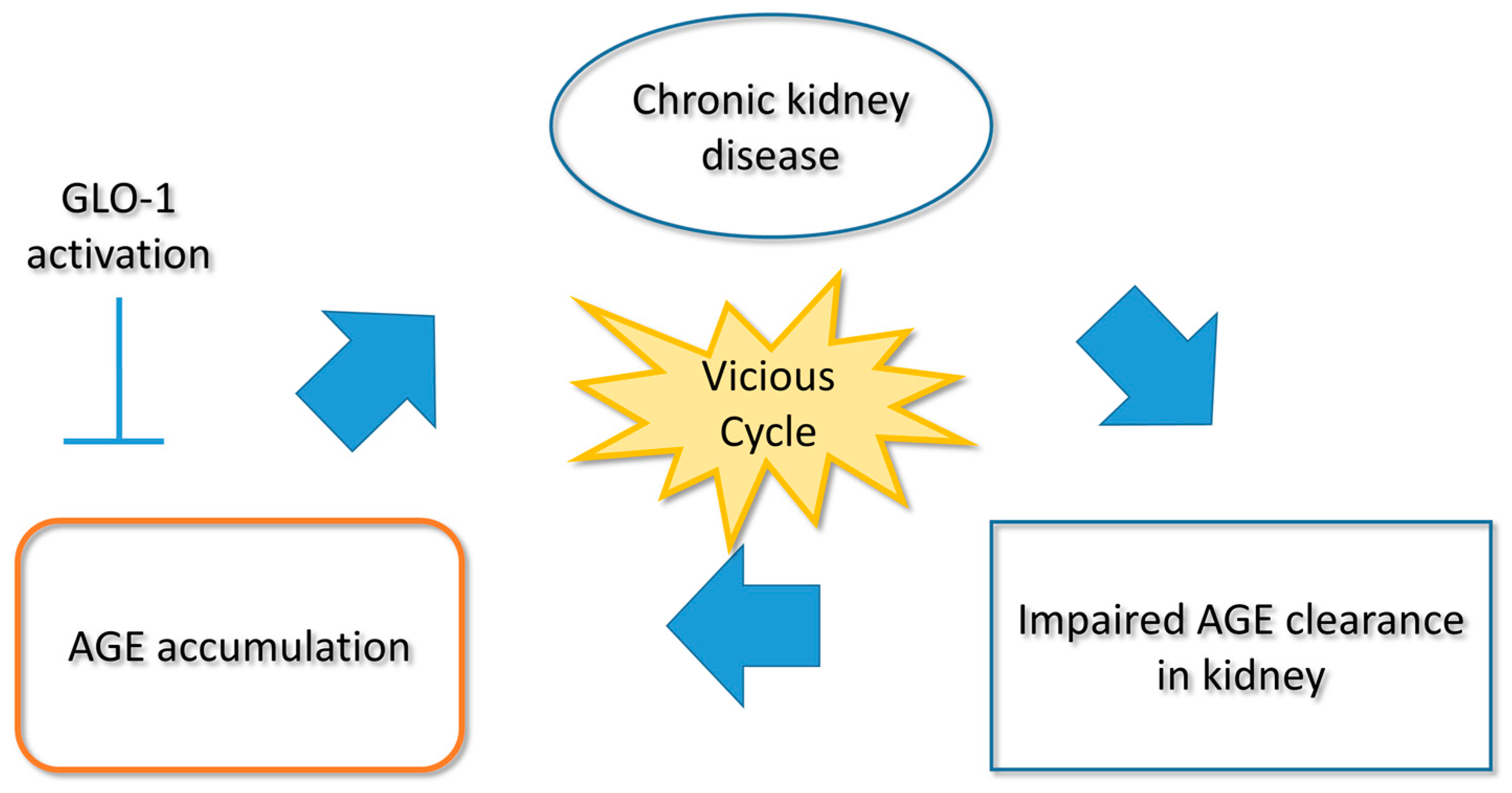
2. Glycative Stress in Pathogenesis of Kidney Disease
2.1. Diabetic Kidney Disease
2.2. Other Non-Diabetic CKD (Chronic Kidney Disease)
3. GLO-1 System in Renal and Vascular Senescence
4. Renal Hypoxia as the Final Common Pathway of Chronic Kidney Disease
5. Cross-Talk between Glycative Stress, ER (Endoplasmic Reticulum) Stress and Hypoxia
6. Intervention in Renal Glycative Stress
6.1. Activating GLO-1
6.2. Suppression of AGE Accumulation and Reduction of AGE Production
7. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- GFR Decreased. Chapter 1: Definition and classification of CKD. Kidney Int. Suppl. 2013, 3, 19–62. [Google Scholar]
- Inagi, R. Glycative stress and glyoxalase in kidney disease and aging. Biochem. Soc. Trans. 2014, 42, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Nikolic-Paterson, D.J.; Wang, S.; Lan, H.Y. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int. Suppl. 2014, 4, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Ishimoto, Y.; Nangaku, M. Proteostasis in endoplasmic reticulum—New mechanisms in kidney disease. Nat. Rev. Nephrol. 2014, 10, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Ueda, Y.; Horie, K.; Nangaku, M.; Tanaka, S.; de Strihou, C.V.Y.; Kurokawa, K. Renal catabolism of advanced glycation end products: The fate of pentosidine. Kidney Int. 1998, 53, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Schlotterer, A.; Kukudov, G.; Bozorgmehr, F.; Hutter, H.; Du, X.; Oikonomou, D.; Ibrahim, Y.; Pfisterer, F.; Rabbani, N.; Thornalley, P.; et al. C. Elegans as model for the study of high glucose-mediated life span reduction. Diabetes 2009, 58, 2450–2456. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 963–975. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, K.; Cai, G.Y.; Chen, X.M.; Yang, J.R.; Lin, L.R.; Yang, J.; Huo, B.G.; Zhan, J.; He, Y.N. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal. 2014, 26, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Nathan, D.M.; Cleary, P.A.; Backlund, J.Y.; Genuth, S.M.; Lachin, J.M.; Orchard, T.J.; Raskin, P.; Zinman, B. Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study Research Group. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 2005, 353, 2643–2653. [Google Scholar] [PubMed]
- Chalmers, J.; Cooper, M.E. UKPDS and the legacy effect. N. Engl. J. Med. 2008, 359, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A. Hypothesis: The “metabolic memory”, the new challenge of diabetes. Diabetes Res. Clin. Pract. 2009, 86, S2–S6. [Google Scholar] [CrossRef]
- Stratton, I.M.; Adler, A.I.; Neil, H.A.; Matthews, D.R.; Manley, S.E.; Cull, C.A.; Hadden, D.; Turner, R.C.; Holman, R.R. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): Prospective observational study. BMJ 2000, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar]
- American Diabetes Association. The absence of a glycemic threshold for the development of long-term complications: The perspective of the diabetes control and complications trial. Diabetes 1996, 45, 1289–1298. [Google Scholar]
- Monnier, V.M.; Bautista, O.; Kenny, D.; Sell, D.R.; Fogarty, J.; Dahms, W.; Cleary, P.A.; Lachin, J.; Genuth, S. Skin collagen glycation, glycoxidation, and crosslinking are lower in subjects with long-term intensive versus conventional therapy of type 1 diabetes: Relevance of glycated collagen products versus HbA1c as markers of diabetic complications. DCCT Skin Collagen Ancillary Study Group. Diabetes Control and Complications Trial. Diabetes 1999, 48, 870–880. [Google Scholar] [PubMed]
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 2014, 63, 50–52. [Google Scholar] [CrossRef] [PubMed]
- Peculis, R.; Konrade, I.; Skapare, E.; Fridmanis, D.; Nikitina-Zake, L.; Lejnieks, A.; Pirags, V.; Dambrova, M.; Klovins, J. Identification of glyoxalase 1 polymorphisms associated with enzyme activity. Gene 2013, 515, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Karachalias, N.; Babaei-Jadidi, R.; Ahmed, N.; Thornalley, P.J. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem. Soc. Trans. 2003, 31, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Østergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [PubMed]
- Kaida, Y.; Fukami, K.; Matsui, T.; Higashimoto, Y.; Nishino, Y.; Obara, N.; Nakayama, Y.; Ando, R.; Toyonaga, M.; Ueda, S.; et al. DNA aptamer raised against ages blocks the progression of experimental diabetic nephropathy. Diabetes 2013, 62, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhen, J.; Zhang, C.; Wan, Q.; Liu, G.; Wei, X.; Zhang, Y.; Wang, Z.; Han, H.; et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int. 2014, 86, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.P.; Chen, C.; Hao, J.; Huang, J.Y.; Liu, P.Q.; Huang, H.Q. AGEs-RAGE system down-regulates Sirt1 through the ubiquitin-proteasome pathway to promote FN and TGF-β1 expression in male rat glomerular mesangial cells. Endocrinology 2015, 156, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Shen, T.T.; Chen, R.H.; Wu, H.L.; Wang, Y.J.; Deng, J.K.; Chen, Q.H.; Pan, Q.; Huang Fu, C.M.; Tao, J.L.; et al. Autophagy-lysosome pathway in renal tubular epithelial cells is disrupted by advanced glycation end products in diabetic nephropathy. J. Biol. Chem. 2015, 290, 20499–20510. [Google Scholar] [CrossRef] [PubMed]
- Shechter, P.; Boner, G.; Rabkin, R. Tubular cell protein degradation in early diabetic renal hypertrophy. J. Am. Soc. Nephrol. 1994, 4, 1582–1587. [Google Scholar] [PubMed]
- Osicka, T.M.; Houlihan, C.A.; Chan, J.G.; Jerums, G.; Comper, W.D. Albuminuria in patients with type 1 diabetes is directly linked to changes in the lysosome-mediated degradation of albumin during renal passage. Diabetes 2000, 49, 1579–1584. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Goldberg, I.J.; Susztak, K. The evolving understanding of the contribution of lipid metabolism to diabetic kidney disease. Curr. Diabetes Rep. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Baragetti, I.; Norata, G.D.; Sarcina, C.; Baragetti, A.; Rastelli, F.; Buzzi, L.; Grigore, L.; Garlaschelli, K.; Pozzi, C.; Catapano, A.L. -374 T/A RAGE polymorphism is associated with chronic kidney disease progression in subjects affected by nephrocardiovascular disease. PLoS ONE 2013, 8, e60089. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R.; Yamamoto, Y.; Nangaku, M.; Usuda, N.; Okamato, H.; Kurokawa, K.; van Ypersele de Strihou, C.V.Y.; Yamamoto, H.; Miyata, T. A severe diabetic nephropathy model with early development of nodule-like lesions induced by megsin overexpression in RAGE/iNOS transgenic mice. Diabetes 2006, 55, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Humpert, P.M.; Nawroth, P.P.; Bierhaus, A. Reactive metabolites and AGE/RAGE-mediated cellular dysfunction affect the aging process: A mini-review. Gerontology 2011, 57, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [PubMed]
- Kihm, L.P.; Wibisono, D.; Müller-Krebs, S.; Pfisterer, F.; Morath, C.; Gross, M.L.; Morcos, M.; Seregin, Y.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE expression in the human peritoneal membrane. Nephrol. Dial. Transplant. 2008, 23, 3302–3306. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Striker, L.J.; Teichberg, S.; Fuh, H.; Li, Y.M.; Steffes, M. Advanced glycation end products induce glomerular sclerosis and albuminuria in normal rats. Proc. Natl. Acad. Sci. USA 1994, 91, 11704–11708. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, M.D.; Bach, L.A.; Forbes, J.M.; Nikolic-Paterson, D.; McRobert, A.; Thallas, V.; Atkins, R.C.; Osicka, T.; Jerums, G.; Cooper, M.E. Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 2001, 108, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to age in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.Y.; Yu, Q.; Fang, W.; Uribarri, J.; He, J.C. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007, 72, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic toxicity of advanced glycation end products in CKD. J. Am. Soc. Nephrol. 2016, 27, 354–370. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, T.; Nangaku, M.; Kojima, I.; Nagai, R.; Ingelfinger, J.R.; Miyata, T.; Fujita, T.; Inagi, R. Glyoxalase I overexpression ameliorates renal ischemia-reperfusion injury in rats. Am. J. Physiol. Ren. Physiol. 2009, 296, F912–F921. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Inagi, R.; Miyata, T.; Nagai, R.; Arai, M.; Miyashita, M.; Itokawa, M.; Fujita, T.; Nangaku, M. Glyoxalase I retards renal senescence. Am. J. Pathol. 2011, 179, 2810–2821. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; de Jong, P.E.; Coresh, J.; El Nahas, M.; Astor, B.C.; Matsushita, K.; Gansevoort, R.T.; Kasiske, B.L.; Eckardt, K.U. The definition, classification, and prognosis of chronic kidney disease: A KDIGO Controversies Conference report. Kidney Int. 2011, 80, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Murray, A.M.; Li, S.; Herzog, C.A.; McBean, A.M.; Eggers, P.W.; Collins, A.J. Chronic kidney disease and the risk for cardiovascular disease, renal replacement, and death in the united states medicare population, 1998 to 1999. J. Am. Soc. Nephrol. 2005, 16, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Shirakawa, J.; Nagai, R.; Miyata, T.; Nagano, T.; et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 2014, 13, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Mimura, I.; Nangaku, M. The suffocating kidney: Tubulointerstitial hypoxia in end-stage renal disease. Nat. Rev. Nephrol. 2010, 6, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Yoshihara, T.; Kamiya, M.; Mimura, I.; Fujikura, D.; Masuda, T.; Kikuchi, R.; Takahashi, I.; Urano, Y.; Tobita, S.; et al. Quantitating intracellular oxygen tension in vivo by phosphorescence lifetime measurement. Sci. Rep. 2015, 5, 17838. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Eto, N.; Nangaku, M. Inflammation and hypoxia linked to renal injury by CCAAT/enhancer-binding protein δ. Kidney Int. 2015, 88, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Cai, W.; Peppa, M.; Goodman, S.; Ferrucci, L.; Striker, G.; Vlassara, H. Circulating glycotoxins and dietary advanced glycation endproducts: Two links to inflammatory response, oxidative stress, and aging. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Inagi, R. RAGE and glyoxalase in kidney disease. Glycoconj. J. 2016, 33, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Nangaku, M.; Tanaka, T.; Iwawaki, T.; Inagi, R. Endoplasmic reticulum stress signal impairs erythropoietin production: A role for ATF4. Am. J. Physiol. Cell Physiol. 2013, 304, C342–C353. [Google Scholar] [CrossRef] [PubMed]
- Nangaku, M.; Mimura, I.; Yamaguchi, J.; Higashijima, Y.; Wada, T.; Tanaka, T. Role of uremic toxins in erythropoiesis-stimulating agent resistance in chronic kidney disease and dialysis patients. J. Ren. Nutr. 2015, 25, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Park, J.E.; Jung, K.H.; Jeon, D.; Lim, J.Y.; Lee, S.K.; Kim, M.; Roh, J.K. Erythropoietin improves memory function with reducing endothelial dysfunction and amyloid-β burden in Alzheimer’s disease models. J. Neurochem. 2012, 120, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, L.; Chen, T.; Liu, Z.; Liu, H.; Li, Z. Erythropoietin attenuates advanced glycation endproducts-induced toxicity of schwann cells in vitro. Neurochem. Res. 2015, 40, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, I.; Ruster, C.; Franke, S.; Liebisch, M.; Wolf, G. Erythropoietin ameliorates podocyte injury in advanced diabetic nephropathy in the db/db mouse. Am. J. Physiol. Ren. Physiol. 2013, 305, F911–F918. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Wang, B.; Zhang, X.F.; Ma, Y.P.; Liu, J.D.; Wang, X.Z. Contribution of receptor for advanced glycation end products to vasculature-protecting effects of exercise training in aged rats. Eur. J. Pharmacol. 2014, 741, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, E.; Murata, Y.; Sakamoto, M.; Horikawa, E. Effects of exercise on the nephron of Goto-Kakizaki rats: Morphological, and advanced glycation end-products and inducible nitric oxide synthase immunohistochemical analyses. J. Diabetes Complicat. 2015, 29, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Zhu, X.; Zhang, L.; Lu, Q.; Wang, J.Y.; Zhang, F.; Guo, H.; Yin, J.L.; Yin, X.X. Up-regulation of glyoxalase 1 by mangiferin prevents diabetic nephropathy progression in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2013, 721, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.M.; Suh, K.S.; Rhee, S.Y.; Kim, Y.S. Sciadopitysin alleviates methylglyoxal-mediated glycation in osteoblastic MC3T3-E1 cells by enhancing glyoxalase system and mitochondrial biogenesis. Free Radic. Res. 2014, 48, 729–739. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Cheng, Y.Q.; Liu, X.L.; Hao, Y.C.; Li, Y.; Zhu, X.; Zhang, F.; Yin, X.X. Mangiferin upregulates glyoxalase 1 through activation of Nrf2/are signaling in central neurons cultured with high glucose. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.S.; Tauchi, M.; Katsuoka, F.; Flanders, K.C.; Liby, K.T.; Honda, T.; Gribble, G.W.; Johnson, D.A.; Johnson, J.A.; Burton, N.C.; et al. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol. Cancer Ther. 2007, 6, 154–162. [Google Scholar] [CrossRef]
- Pergola, P.E.; Krauth, M.; Huff, J.W.; Ferguson, D.A.; Ruiz, S.; Meyer, C.J.; Warnock, D.G. Effect of bardoxolone methyl on kidney function in patients with T2D and stage 3b–4 CKD. Am. J. Nephrol. 2011, 33, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Pergola, P.E.; Raskin, P.; Toto, R.D.; Meyer, C.J.; Huff, J.W.; Grossman, E.B.; Krauth, M.; Ruiz, S.; Audhya, P.; Christ-Schmidt, H.; et al. Bardoxolone methyl and kidney function in CKD with type 2 diabetes. N. Engl. J. Med. 2011, 365, 327–336. [Google Scholar] [CrossRef] [PubMed]
- De Zeeuw, D.; Akizawa, T.; Audhya, P.; Bakris, G.L.; Chin, M.; Christ-Schmidt, H.; Goldsberry, A.; Houser, M.; Krauth, M.; Lambers Heerspink, H.J.; et al. Bardoxolone methyl in type 2 diabetes and stage 4 chronic kidney disease. N. Engl. J. Med. 2013, 369, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Semba, R.D.; Gebauer, S.K.; Baer, D.J.; Sun, K.; Turner, R.; Silber, H.A.; Talegawkar, S.; Ferrucci, L.; Novotny, J.A. Dietary intake of advanced glycation end products did not affect endothelial function and inflammation in healthy adults in a randomized controlled trial. J. Nutr. 2014, 144, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.E.; Dordevic, A.L.; Tan, S.M.; Ryan, L.; Coughlan, M.T. Dietary advanced glycation end products and risk factors for chronic disease: A systematic review of randomised controlled trials. Nutrients 2016, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; He, J.C. The low age diet: A neglected aspect of clinical nephrology practice? Nephron 2015, 130, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Kakuta, T.; Tanaka, R.; Hyodo, T.; Suzuki, H.; Kanai, G.; Nagaoka, M.; Takahashi, H.; Hirawa, N.; Oogushi, Y.; Miyata, T.; et al. Effect of sevelamer and calcium-based phosphate binders on coronary artery calcification and accumulation of circulating advanced glycation end products in hemodialysis patients. Am. J. Kidney Dis. 2011, 57, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H.; Uribarri, J.; Cai, W.; Goodman, S.; Pyzik, R.; Post, J.; Grosjean, F.; Woodward, M.; Striker, G.E. Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin. J. Am. Soc. Nephrol. 2012, 7, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Yubero-Serrano, E.M.; Woodward, M.; Poretsky, L.; Vlassara, H.; Striker, G.E. Effects of sevelamer carbonate on advanced glycation end products and antioxidant/pro-oxidant status in patients with diabetic kidney disease. Clin. J. Am. Soc. Nephrol. 2015, 10, 759–766. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003, 419, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Baynes, J.W.; Thorpe, S.R.; Cooper, M.E. The role of AGEs and AGE inhibitors in diabetic cardiovascular disease. Curr. Drug Targets 2005, 6, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Booth, A.A.; Khalifah, R.G.; Hudson, B.G. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: Comparison with aminoguanidine. Biochem. Biophys. Res. Commun. 1996, 220, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Babaei-Jadidi, R.; Karachalias, N.; Ahmed, N.; Battah, S.; Thornalley, P.J. Prevention of incipient diabetic nephropathy by high-dose thiamine and benfotiamine. Diabetes 2003, 52, 2110–2120. [Google Scholar] [CrossRef] [PubMed]
- Kihm, L.P.; Müller-Krebs, S.; Klein, J.; Ehrlich, G.; Mertes, L.; Gross, M.L.; Adaikalakoteswari, A.; Thornalley, P.J.; Hammes, H.P.; Nawroth, P.P.; et al. Benfotiamine protects against peritoneal and kidney damage in peritoneal dialysis. J. Am. Soc. Nephrol. 2011, 22, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Muller-Krebs, S.; Nissle, K.; Tsobaneli, J.; Zeier, M.; Kihm, L.P.; Kender, Z.; Fleming, T.; Nawroth, P.P.; Reiser, J.; Schwenger, V. Effect of benfotiamine in podocyte damage induced by peritoneal dialysis fluid. Front. Med. 2015, 2, 10. [Google Scholar]
- Booth, A.A.; Khalifah, R.G.; Todd, P.; Hudson, B.G. In vitro kinetic studies of formation of antigenic advanced glycation end products (AGEs). Novel inhibition of post-Amadori glycation pathways. J. Biol. Chem. 1997, 272, 5430–5437. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, T.P.; Alderson, N.L.; Arrington, D.D.; Beattie, R.J.; Basgen, J.M.; Steffes, M.W.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat. Kidney Int. 2002, 61, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Golegaonkar, S.; Tabrez, S.S.; Pandit, A.; Sethurathinam, S.; Jagadeeshaprasad, M.G.; Bansode, S.; Sampathkumar, S.G.; Kulkarni, M.J.; Mukhopadhyay, A. Rifampicin reduces advanced glycation end products and activates DAF-16 to increase lifespan in Caenorhabditis elegans. Aging Cell 2015, 14, 463–473. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirakawa, Y.; Inagi, R. Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis. Int. J. Mol. Sci. 2017, 18, 174. https://doi.org/10.3390/ijms18010174
Hirakawa Y, Inagi R. Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis. International Journal of Molecular Sciences. 2017; 18(1):174. https://doi.org/10.3390/ijms18010174
Chicago/Turabian StyleHirakawa, Yosuke, and Reiko Inagi. 2017. "Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis" International Journal of Molecular Sciences 18, no. 1: 174. https://doi.org/10.3390/ijms18010174
APA StyleHirakawa, Y., & Inagi, R. (2017). Glycative Stress and Its Defense Machinery Glyoxalase 1 in Renal Pathogenesis. International Journal of Molecular Sciences, 18(1), 174. https://doi.org/10.3390/ijms18010174