
Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Integrin Activation Elicits Proliferative and Survival Signaling
1.2. Integrin Internalization and Membrane Traffic
1.3. Integrins Control Cell Adhesion and Migration
1.4. Integrins and Cancer
2. Metabolic Signals and Alterations in Cancer
2.1. Metabolic Control of AMP-Activated Protein Kinase (AMPK) and Its Role in Cancer
2.2. Mammalian Target of Rapamycin (mTOR) Integrates Amino Acid Sensing and Mitogenic Signaling
2.3. Hypoxia Sensing by HIF1 Coordinates Metabolic Adaptation
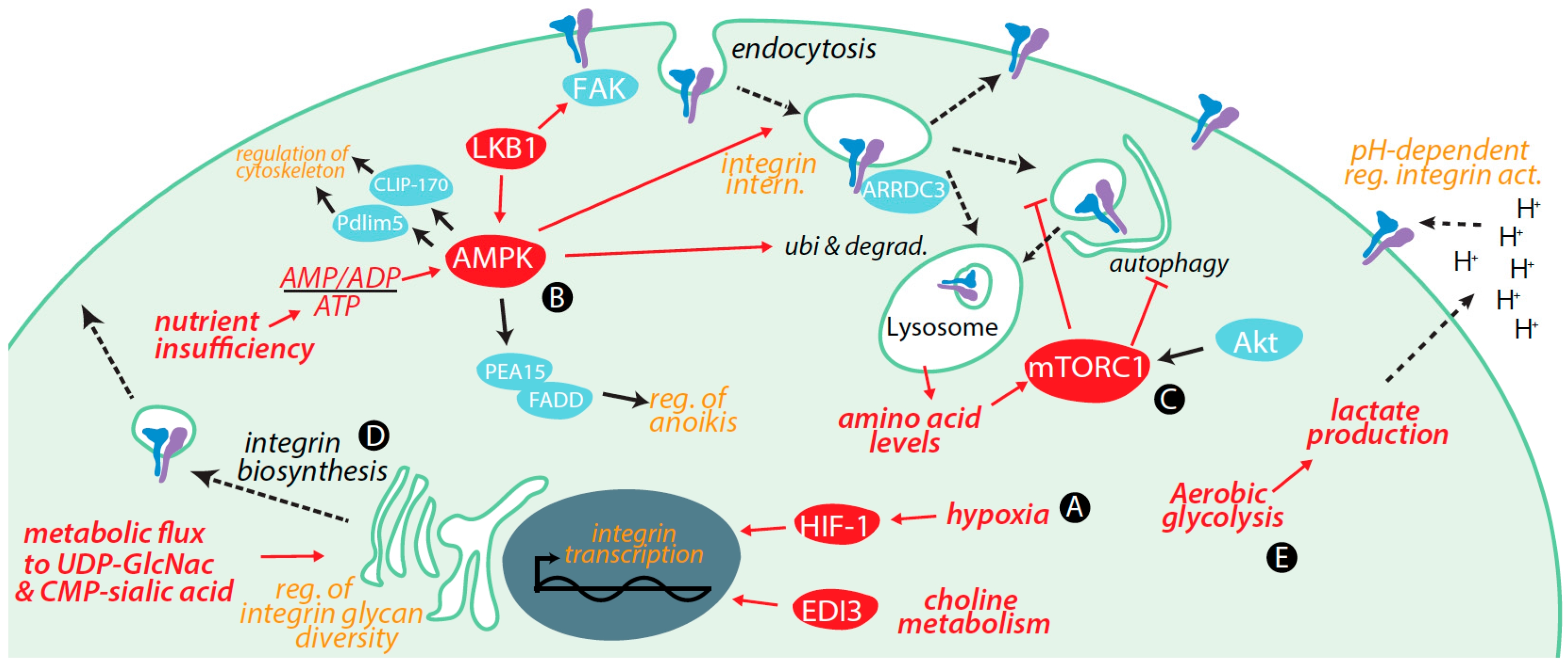
3. Regulation of Integrins by Metabolic Cues and Signaling
3.1. Transcriptional Control of Integrin Expression by Metabolic Signals
3.2. Control of Integrin Membrane Traffic by Metabolic Signals
3.3. Control of Integrin Degradation by Metabolic Cues
3.4. Further Control of Integrin Expression by Metabolic Signals
3.5. Integrin Glycosylation Is Controlled by Metabolic Inputs
3.6. Metabolic Signals Control Cell Migration and Adhesion
3.7. Metabolic Cues Control Integrin Signaling
3.8. Metabolic Control of Integrins via Alterations in Tumour Microenvironment
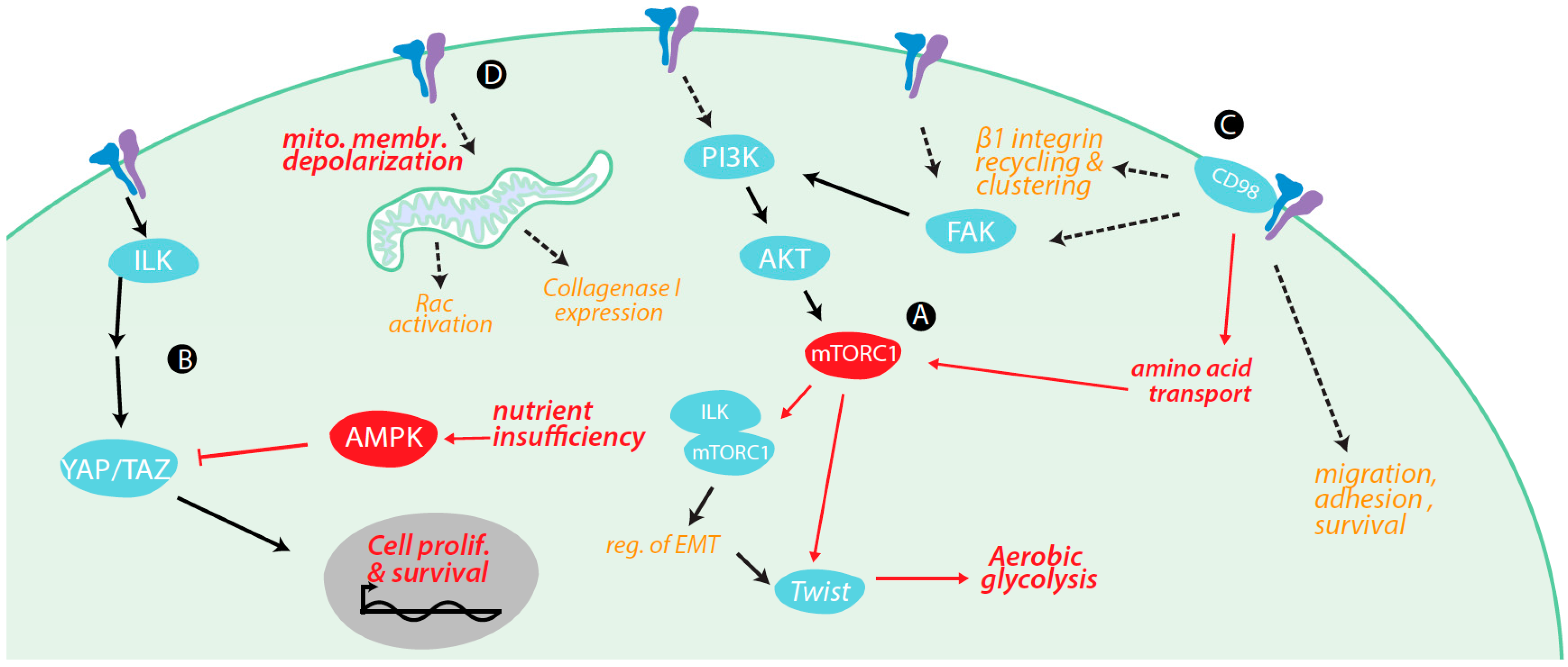
4. Regulation of Metabolism and Metabolically-Regulated Signals by Integrins
4.1. Integrin Signaling Cross-Talk with Metabolic Signaling
4.2. Integrin Signaling Controls Metabolic Pathways
4.3. Integrin Control of Nutrient and Metabolite Transporters
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 4EBP1 | Eukaryotic translation initiation factor 4E-binding protein 1 |
| AMPK | AMP-activated protein kinase |
| ARRDC3 | Arrestin Domain Containing 3 |
| CMP-sialic acid | Cytidine-5′-monophospho-N-acetylneuraminic acid |
| CPT1C | Carnitine palmitoyltransferase 1C |
| DOAJ | Directory of open access journals |
| ECM | Extracellular matrix |
| EDI3 | Endometrial carcinoma differential 3 |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| FAK | Focal adhesion kinase |
| FA | Focal adhesion |
| FADD | Fas-Associated protein with Death Domain |
| GAP | GTPase Activating Protein |
| GBM | Glioblastoma multiforme |
| GEF | Guanyl Exchange Factor |
| GLUT1 | Facilitative glucose transporter 1 |
| GnT-V | β1-6-N-Acetylglucosaminyltransferase V |
| HIF | Hypoxia Inducible Factor |
| HMEC | Human mammary epithelial cells |
| ILK | Integrin linked kinase |
| JNK | c-Jun N-terminal kinase |
| LAT1/2 | L-Type Amino Acid Transporter 1/2 |
| LKB1 | Liver Kinase B1 |
| MAPK | Mitogen-activated protein kinase |
| MCT | Monocarboxylate transporter |
| MDPI | Multidisciplinary Digital Publishing Institute |
| mTOR | Mammalian Target of Rapamycin |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MYPT1 | Myosin Phosphatase Target Subunit 1 |
| O-GlcNAc | O-Linked β N-acetylglucosamine |
| OGT | O-GlcNAc Transferase |
| PFK | Phosphofructokinase |
| PI3K | Phosphatidylinotiol-3-kinase |
| PIP3 | Phosphatidylinositol-3,4,5-trisphosphate |
| PK | Phosphofructokinase |
| PTEN | Phosphatase and tensin homolog |
| RGD | Arginyl-glycyl-aspartic acid (motif) |
| ROS | Reactive oxygen species |
| RHEB | Ras homologue enriched in brain |
| TAZ | Transcriptional coactivator with PDZ-binding motif, also known as WWTR1 |
| TSC | Tubular sclerosis complex |
| SREBP | Sterol regulatory element-binding transcription factor |
| STGal6 I | β-Galactoside α2,6-sialyltransferase I |
| UDP-GlcNAc | Uridine 5′-diphospho-N-acetylglucosamine |
| YAP | Yes-associated protein |
References
- Ganguly, K.K.; Pal, S.; Moulik, S.; Chatterjee, A. Integrins and metastasis. Cell Adhes. Migr. 2013, 7, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Harburger, D.S.; Calderwood, D.A. Integrin signaling at a glance. J. Cell Sci. 2008, 122, 1472. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef] [PubMed]
- De Franceschi, N.; Hamidi, H.; Alanko, J.; Sahgal, P.; Ivaska, J. Integrin traffic—The update. J. Cell Sci. 2015, 128, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Partridge, A.W.; Liu, S.; Kim, S.; Bowie, J.U.; Ginsberg, M.H. Transmembrane domain helix packing stabilizes integrin αIIbβ3 in the low affinity state. J. Biol. Chem. 2005, 280, 7294–7300. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. Integrins bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Wegener, K.L.; Partridge, A.W.; Han, J.; Pickford, A.R.; Liddington, R.C.; Ginsberg, M.H.; Campbell, I.D. Structural basis of integrin activation by Talin. Cell 2007, 128, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Ivaska, J.; Heino, J. Cooperation between integrins and growth factor receptors in signaling and endocytosis. Annu. Rev. Cell Dev. Biol. 2011, 27, 291–320. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Giancotti, F.G. Integrin signaling furing tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, R.T.; Lange, A.; Fässler, R. How ILK and kindlins cooperate to orchestrate integrin signaling. Curr. Opin. Cell Biol. 2009, 21, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Arias-Salgado, E.G.; Lizano, S.; Sarkar, S.; Brugge, J.S.; Ginsberg, M.H.; Shattil, S.J. Src kinase activation by direct interaction with the integrin β cytoplasmic domain. Proc. Natl. Acad. Sci. USA 2003, 100, 13298–13302. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Appeddu, P.A.; Isoda, H.; Guan, J.L. Phosphorylation of tyrosine 397 in focal adhesion kinase is required for binding phosphatidylinositol 3-kinase. J. Biol. Chem. 1996, 271, 26329–26334. [Google Scholar] [PubMed]
- Xia, H.; Nho, R.S.; Kahm, J.; Kleidon, J.; Henke, C.A. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a β1 integrin viability signaling pathway. J. Biol. Chem. 2004, 279, 33024–33034. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.L.; Reginato, M.J.; Paulus, J.K.; Sgroi, D.C.; Labaer, J.; Brugge, J.S. G1/S cell cycle arrest provides anoikis resistance through Erk-mediated Bim suppression. Mol. Cell. Biol. 2005, 25, 5282–5291. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Clifford, J.L.; Chung, J. Crosstalk between integrin and receptor tyrosine kinase signaling in breast carcinoma progression. BMB Rep. 2010, 43, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Streuli, C.H.; Akhtar, N. Signal co-operation between integrins and other receptor systems. Biochem. J. 2009, 418, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Pylayeva, Y.; Pepe, A.; Yoshioka, T.; Muller, W.J.; Inghirami, G.; Giancotti, F.G. β4 Integrin amplifies ErbB2 signaling to promote mammary tumourigenesis. Cell 2006, 126, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Cabodi, S.; Morello, V.; Masi, A.; Cicchi, R.; Broggio, C.; Distefano, P.; Brunelli, E.; Silengo, L.; Pavone, F.; Arcangeli, A.; et al. Convergence of integrins and EGF receptor signaling via PI3K/Akt/FoxO pathway in early gene Egr-1 expression. J. Cell. Physiol. 2009, 218, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Moro, L.; Venturino, M.; Bozzo, C.; Silengo, L.; Altruda, F.; Beguinot, L.; Tarone, G.; Defilippi, P. Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. EMBO J. 1998, 17, 6622–6632. [Google Scholar] [CrossRef] [PubMed]
- Morello, V.; Cabodi, S.; Sigismund, S.; Camacho-Leal, M.P.; Repetto, D.; Volante, M.; Papotti, M.; Turco, E.; Defilippi, P. β1 integrin controls EGFR signaling and tumourigenic properties of lung cancer cells. Oncogene 2011, 30, 4087–4096. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Juliano, R.L. The α5β1 integrin selectively enhances epidermal growth factor signaling to the phosphatidylinositol-3-kinase/Akt pathway in intestinal epithelial cells. Biochim. Biophys. Acta 2002, 1542, 23–31. [Google Scholar] [CrossRef]
- Delos Santos, R.C.; Garay, C.; Antonescu, C.N. Charming neighborhoods on the cell surface: Plasma membrane microdomains regulate receptor tyrosine kinase signaling. Cell Signal. 2015, 27, 1963–1976. [Google Scholar] [CrossRef] [PubMed]
- Ricono, J.M.; Huang, M.; Barnes, L.A.; Lau, S.K.; Weis, S.M.; Schlaepfer, D.D.; Hanks, S.K.; Cheresh, D.A. Specific cross-talk between epidermal growth factor receptor and integrin αvβ5 promotes carcinoma cell invasion and metastasis. Cancer Res. 2009, 69, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Balanis, N.; Yoshigi, M.; Wendt, M.K.; Schiemann, W.P.; Carlin, C.R. Integrin-EGF receptor cross-talk activates p190RhoGAP in mouse mammary gland epithelial cells. Mol. Biol. Cell 2011, 22, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Vachon, H.P. Integrin signaling, cell survival, and anoikis: Distinctions, differences, and differentiation. J. Signal. Transduct. 2011, 2011, 738137. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.; Norman, J. Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 2008, 18, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Bridgewater, R.E.; Norman, J.C.; Caswell, P.T. Integrin trafficking at a glance. J. Cell Sci. 2012, 125, 3695–3701. [Google Scholar] [CrossRef] [PubMed]
- Pellinen, T.; Tuomi, S.; Arjonen, A.; Wolf, M.; Edgren, H.; Meyer, H.; Grosse, R.; Kitzing, T.; Rantala, J.K.; Kallioniemi, O.; et al. Integrin trafficking regulated by Rab21 is necessary for cytokinesis. Dev. Cell 2008, 15, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Ezratty, E.J.; Bertaux, C.; Marcantonio, E.E.; Gundersen, G.G. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J. Cell Biol. 2009, 187, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Teckchandani, A.; Mulkearns, E.E.; Randolph, T.W.; Toida, N.; Cooper, J.A. The clathrin adaptor Dab2 recruits EH domain scaffold proteins to regulate integrin β1 endocytosis. Mol. Biol. Cell 2012, 23, 2905–2916. [Google Scholar] [CrossRef] [PubMed]
- Teckchandani, A.; Toida, N.; Goodchild, J.; Henderson, C.; Watts, J.; Wollscheid, B.; Cooper, J.A. Quantitative proteomics identifies a Dab2/integrin module regulating cell migration. J. Cell Biol. 2009, 186, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Sottile, J. Caveolin-1-dependent β1 integrin endocytosis is a critical regulator of fibronectin turnover. J. Cell Sci. 2008, 121, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Fujioka, Y.; de Pereda, J.M.; Garcia-Alvarez, B.; Nakamoto, T.; Margolis, B.; McGlade, C.J.; Liddington, R.C.; Ginsberg, M.H. Integrin β cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc. Natl. Acad. Sci. USA 2003, 100, 2272–2277. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-W.; Luo, R.; Jian, X.; Randazzo, P.A. The Arf6 GTPase-activating proteins ARAP2 and ACAP1 define distinct endosomal compartments that regulate integrin α5β1 traffic. J. Biol. Chem. 2014, 289, 30237–30248. [Google Scholar] [CrossRef] [PubMed]
- Dozynkiewicz, M.A.; Jamieson, N.B.; Macpherson, I.; Grindlay, J.; van den Berghe, P.V.E.; von Thun, A.; Morton, J.P.; Gourley, C.; Timpson, P.; Nixon, C.; et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev. Cell 2012, 22, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Powelka, A.M.; Sun, J.; Li, J.; Gao, M.; Shaw, L.M.; Sonnenberg, A.; Hsu, V.W. Stimulation-Dependent Recycling of Integrin β1 Regulated by Arf6 and Rab11. Traffic 2004, 5, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Santy, L.C. Phosphoinositide specificity determines which cytohesins regulate β1 integrin recycling. J. Cell Sci. 2012, 125, 3195–3201. [Google Scholar] [CrossRef] [PubMed]
- Pellinen, T.; Arjonen, A.; Vuoriluoto, K.; Kallio, K.; Fransen, J.A.M.; Ivaska, J. Small GTPase Rab21 regulates cell adhesion and controls endosomal traffic of β1-integrins. J. Cell Biol. 2006, 173, 767–780. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.; Pang, X.; Lou, J.; Zhou, Q.; Zhang, K.; Ma, J.; Li, J.; Sun, F.; Hsu, V.W. Mechanistic insights into regulated cargo binding by ACAP1 protein. J. Biol. Chem. 2012, 287, 28675–28685. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Norman, J.C. Integrin trafficking and the control of cell migration. Traffic 2006, 7, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK—Src signaling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Regen, C.M.; Horwitz, A.F. Dynamics of β1 integrin-mediated adhesive contacts in motile fibroblasts. J. Cell Biol. 1992, 119, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Smilenov, L.B.; Mikhailov, A.; Pelham, R.J.; Marcantonio, E.E.; Gundersen, G.G. Focal adhesion motility revealed in stationary fibroblasts. Science 1999, 286, 1172–1174. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Elevated levels of the α5β1 fibronectin receptor suppress the transformed phenotype of Chinese hamster ovary cells. Cell 1990, 60, 849–859. [Google Scholar] [CrossRef]
- Varner, J.A.; Emerson, D.A.; Juliano, R.L. Integrin α5β1 expression negatively regulates cell growth: Reversal by attachment to fibronectin. Mol. Biol. Cell 1995, 6, 725–740. [Google Scholar] [CrossRef] [PubMed]
- Desgrosellier, J.S.; Barnes, L.A.; Shields, D.J.; Huang, M.; Lau, S.K.; Prévost, N.; Tarin, D.; Shattil, S.J.; Cheresh, D.A. An integrin αvβ3–c-Src oncogenic unit promotes anchorage-independence and tumour progression. Nat. Med. 2009, 15, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Vanden Berg, S.R.; Bergers, G.; Berger, M.S.; Pieper, R.O. Integrin β3 overexpression suppresses tumour growth in a human model of gliomagenesis: Implications for the role of β3 overexpression in glioblastoma multiforme. Cancer Res. 2004, 64, 2751–2758. [Google Scholar] [CrossRef] [PubMed]
- Janes, S.M.; Watt, F.M. Switch from αvβ5 to αvβ6 integrin expression protects squamous cell carcinomas from anoikis. J. Cell Biol. 2004, 166, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Spence, H.J.; Parsons, M.; White, D.P.; Clark, K.; Cheng, K.W.; Mills, G.B.; Humphries, M.J.; Messent, A.J.; Anderson, K.I.; et al. Rab25 associates with α5β1 integrin to promote invasive migration in 3D microenvironments. Dev. Cell 2007, 13, 496–510. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Bell, E.S.; Girard, M.; Chaineau, M.; Hamlin, J.N.R.; Daubaras, M.; Monast, A.; Park, M.; Hodgson, L.; McPherson, P.S. DENND2B activates Rab13 at the leading edge of migrating cells and promotes metastatic behavior. J. Cell Biol. 2015, 208, 629–648. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kugler, M.C.; Wei, Y.; Kim, K.K.; Li, X.; Brumwell, A.N.; Chapman, H.A. Integrin α3β1—Dependent β-catenin phosphorylation links epithelial Smad signaling to cell contacts. J. Cell Biol. 2009, 184, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Pursell, B.; Lu, S.; Chang, T.-K.; Mercurio, A.M. Regulation of β4-integrin expression by epigenetic modifications in the mammary gland and during the epithelial-to-mesenchymal transition. J. Cell Sci. 2009, 122, 2473–2480. [Google Scholar] [CrossRef] [PubMed]
- Rolli, M.; Fransvea, E.; Pilch, J.; Saven, A.; Felding-Habermann, B. Activated integrin αvβ3 cooperates with metalloproteinase MMP-9 in regulating migration of metastatic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9482–9487. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Maschler, S.; Wirl, G.; Spring, H.; Bredow, D.V.; Sordat, I.; Beug, H.; Reichmann, E. Tumour cell invasiveness correlates with changes in integrin expression and localization. Oncogene 2005, 24, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Über den Stoffwechsel der Tumouren (On metabolism of tumours). Biochem. Z 1924, 152, 319–344. [Google Scholar]
- Hardie, D.G. AMPK: A key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. 2008, 32 (Suppl. 4), S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, M. Transport of sugars in tumour cell membranes. Biochim. Biophys. Acta 1974, 355, 77–104. [Google Scholar] [PubMed]
- Dombrauckas, J.D.; Santarsiero, B.D.; Mesecar, A.D. Structural basis for tumour pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry 2005, 44, 9417–9429. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W. Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 2013, 13, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Nilsson, R.; Sharma, S.; Madhusudhan, N.; Kitami, T.; Souza, A.L.; Kafri, R.; Kirschner, M.W.; Clish, C.B.; Mootha, V.K. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 2012, 336, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Xia, Y.; Ding, J.; Ye, B.; Zhao, E.; Choi, J.H.; Alptekin, A.; Yan, C.; Dong, Z.; Huang, S.; et al. Transcriptional profiling reveals a common metabolic program in high-risk human neuroblastoma and mouse neuroblastoma sphere-forming cells. Cell Rep. 2016, 17, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Kottakis, F.; Nicolay, B.N.; Roumane, A.; Karnik, R.; Gu, H.; Nagle, J.M.; Boukhali, M.; Hayward, M.C.; Li, Y.Y.; Chen, T.; et al. LKB1 loss links serine metabolism to DNA methylation and tumourigenesis. Nature 2016, 539, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, C.F.; van den Broek, N.J.F.; Mackay, G.M.; Vousden, K.H.; Maddocks, O.D.K. Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 2014, 7, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Woeltje, K.F.; Kuwajima, M.; Foster, D.W. Regulation of ketogenesis and the renaissance of carnitine palmitoyltransferase. Diabetes Metab. Rev. 1989, 5, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Zaugg, K.; Yao, Y.; Reilly, P.T.; Kannan, K.; Kiarash, R.; Mason, J.; Huang, P.; Sawyer, S.K.; Fuerth, B.; Faubert, B.; et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumour growth under conditions of metabolic stress. Genes Dev. 2011, 25, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A cancer hallmark even warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.; Papandreou, I.; Denko, N. Overcoming physiologic barriers to cancer treatment by molecularly targeting the tumour microenvironment. Mol. Cancer Res. 2006, 4, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumours: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar] [PubMed]
- Kato, Y.; Ozawa, S.; Miyamoto, C.; Maehata, Y.; Suzuki, A.; Maeda, T.; Baba, Y. Acidic extracellular microenvironment and cancer. Cancer Cell Int. 2013, 13, 89. [Google Scholar] [CrossRef] [PubMed]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumour microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Romero-Garcia, S.; Moreno-Altamirano, M.M.B.; Prado-Garcia, H.; Sánchez-García, F.J. Lactate contribution to the tumour microenvironment: Mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol. 2016, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Mbeunkui, F.; Johann, D.J. Cancer and the tumour microenvironment: A review of an essential relationship. Cancer Chemother. Pharmacol. 2009, 63, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Mäkelä, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumour suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumour suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.-P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-β is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Sanders, M.J.; Grondin, P.O.; Hegarty, B.D.; Snowden, M.A.; Carling, D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem. J. 2007, 403, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Emerling, B.M.; Weinberg, F.; Snyder, C.; Burgess, Z.; Mutlu, G.M.; Viollet, B.; Budinger, G.R.; Chandel, N.S. Hypoxic activation of AMPK is dependent on mitochondrial ROS but independent of an increase in AMP/ATP ratio. Free Radic. Biol. Med. 2009, 46, 1386–1391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xie, Z.; Dong, Y.; Wang, S.; Liu, C.; Zou, M.H. Identification of nitric oxide as an endogenous activator of the AMP-activated protein kinase in vascular endothelial cells. J. Biol. Chem. 2008, 283, 27452–27461. [Google Scholar] [CrossRef] [PubMed]
- Kubota, N.; Yano, W.; Kubota, T.; Yamauchi, T.; Itoh, S.; Kumagai, H.; Kozono, H.; Takamoto, I.; Okamoto, S.; Shiuchi, T.; et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab. 2007, 6, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Minokoshi, Y.; Kim, Y.-B.; Peroni, O.D.; Fryer, L.G.D.; Müller, C.; Carling, D.; Kahn, B.B. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 2002, 415, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, M.; Kambe, F.; Cao, X.; Lu, X.; Kozaki, Y.; Oiso, Y.; Seo, H. Thyroid hormone activates adenosine 5′-monophosphate-activated protein kinase via intracellular calcium mobilization and activation of calcium/calmodulin-dependent protein kinase kinase-β. Mol. Endocrinol. 2008, 22, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Walkinshaw, D.R.; Sheehan, T.E.; Hood, D.A. Thyroid hormone (T3) rapidly activates p38 and AMPK in skeletal muscle in vivo. J. Appl. Physiol. 2007, 104, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Kola, B.; Hubina, E.; Tucci, S.A.; Kirkham, T.C.; Garcia, E.A.; Mitchell, S.E.; Williams, L.M.; Hawley, S.A.; Hardie, D.G.; Grossman, A.B.; et al. Cannabinoids and ghrelin have both central and peripheral metabolic and cardiac effects via AMP-activated protein kinase. J. Biol. Chem. 2005, 280, 25196–25201. [Google Scholar] [CrossRef] [PubMed]
- Dando, I.; Donadelli, M.; Costanzo, C.; Dalla Pozza, E.; D’Alessandro, A.; Zolla, L.; Palmieri, M. Cannabinoids inhibit energetic metabolism and induce AMPK-dependent autophagy in pancreatic cancer cells. Cell Death Dis. 2013, 4, e664. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1–AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.; Alessi, D.R. LKB1 and AMPK and the cancer-metabolism link—Ten years after. BMC Biol. 2013, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ramsey, M.R.; Hayes, D.N.; Fan, C.; McNamara, K.; Kozlowski, P.; Torrice, C.; Wu, M.C.; Shimamura, T.; Perera, S.A.; et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 2007, 448, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signaling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Perluigi, M.; di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.-L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Binda, M.; Péli-Gulli, M.-P.; Bonfils, G.; Panchaud, N.; Urban, J.; Sturgill, T.W.; Loewith, R.; de Virgilio, C. The Vam6 GEF controls TORC1 by activating the EGO complex. Mol. Cell 2009, 35, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Shimobayashi, M.; Hall, M.N. Multiple amino acid sensing inputs to mTORC1. Cell Res. 2016, 26, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Milkereit, R.; Persaud, A.; Vanoaica, L.; Guetg, A.; Verrey, F.; Rotin, D. LAPTM4b recruits the LAT1-4F2hc Leu transporter to lysosomes and promotes mTORC1 activation. Nat. Commun. 2015, 6, 7250. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.D.; Zhang, Y.-J.; Wei, Y.-H.; Cho, J.-H.; Morris, L.E.; Wang, H.Y.; Zheng, X.F. Rab1A is an mTORC1 activator and a colorectal oncogene. Cancer Cell 2014, 26, 754–769. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Kim, Y.C.; Russell, R.C.; Yu, F.-X.; Park, H.W.; Plouffe, S.W.; Tagliabracci, V.S.; Guan, K.L. Differential regulation of mTORC1 by leucine and glutamine. Science 2015, 347, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Stracka, D.; Jozefczuk, S.; Rudroff, F.; Sauer, U.; Hall, M.N. Nitrogen source activates TOR (target of rapamycin) complex 1 via glutamine and independently of Gtr/Rag proteins. J. Biol. Chem. 2014, 289, 25010–25020. [Google Scholar] [CrossRef] [PubMed]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR Complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. mTORC1-Dependent metabolic reprogramming underlies escape from glycolysis addiction in cancer cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Sataur, A.; Wang, L.; Chen, H.; Simon, M.C. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1 and HIF-2. Mol. Biol. Cell 2007, 18, 4528–4542. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl 4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Jaakkola, P.; Mole, D.R.; Tian, Y.-M.; Wilson, M.I.; Gielbert, J.; Gaskell, S.J.; von Kriegsheim, A.; Hebestreit, H.F.; Mukherji, M.; Schofield, C.J.; et al. Targeting of HIF-α to the von hippel-lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 2001, 292, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Cockman, M.E.; Masson, N.; Mole, D.R.; Jaakkola, P.; Chang, G.-W.; Clifford, S.C.; Maher, E.R.; Pugh, C.W.; Ratcliffe, P.J.; Maxwell, P.H. Hypoxia inducible factor—Binding and ubiquitylation by the von hippel-lindau tumour suppressor protein. J. Biol. Chem. 2000, 275, 25733–25741. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [PubMed]
- Stiehl, D.P.; Wirthner, R.; Koditz, J.; Spielmann, P.; Camenisch, G.; Wenger, R.H. Increased prolyl 4-hydroxylase domain proteins compensate for decreased oxygen levels: Evidence for an autoregulatory oxygen-sensing system. J. Biol. Chem. 2006, 281, 23482–23491. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1α expression and function by the mammalian target of rapamycin. Mol. Cell. Biol. 2002, 22, 7004–7014. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.-H.; Lee, E.-S.; You, H.-J.; Lee, J.W.; Park, J.-W.; Chun, Y.S. Ras-dependent induction of HIF-1α785 via the Raf/MEK/ERK pathway: A novel mechanism of Ras-mediated tumour promotion. Oncogene 2004, 23, 9427–9431. [Google Scholar] [CrossRef] [PubMed]
- Karni, R.; Dor, Y.; Keshet, E.; Meyuhas, O.; Levitzki, A. Activated pp60c-Src leads to elevated hypoxia-inducible factor (HIF)-1α expression under normoxia. J. Biol. Chem. 2002, 277, 42919–42925. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Kimura, N.; Miyazaki, K.; Yabuta, T.; Kumamoto, K.; Takenoshita, S.; Chen, J.; Kobayashi, M.; Hosokawa, M.; Taniguchi, A.; et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc. Natl. Acad. Sci. USA 2004, 101, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Keely, S.; Glover, L.E.; MacManus, C.F.; Campbell, E.L.; Scully, M.M.; Furuta, G.T.; Colgan, S.P. Selective induction of integrin β1 by hypoxia-inducible factor: Implications for wound healing. FASEB J. 2009, 23, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.H.; Park, H.M.; Chung, J.; Lee, C.H.; Park, H.R. Hypoxia-inducible factor-1α mediates oral squamous cell carcinoma invasion via upregulation of α5 integrin and fibronectin. Biochem. Biophys Res. Commun. 2010, 393, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of β2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 101, 10440–10445. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.-H.; Lu, M.-E. Adiponectin increases motility of human prostate cancer cells via adipoR, p38, AMPK, and NF-κB pathways. Prostate 2009, 69, 1781–1789. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.-C.; Shieh, D.-C.; Tong, K.-M.; Chen, C.-P.; Huang, K.C.; Chen, P.C.; Fong, Y.C.; Hsu, H.C.; Tang, C.H. Involvement of AdipoR receptor in adiponectin-induced motility and α2β1 integrin upregulation in human chondrosarcoma cells. Carcinogenesis 2009, 30, 1651–1659. [Google Scholar] [CrossRef] [PubMed]
- Marchan, R.; Lesjak, M.S.; Stewart, J.D.; Winter, R.; Seeliger, J.; Hengstler, J.G. Choline-releasing glycerophosphodiesterase EDI3 links the tumour metabolome to signaling network activities. Cell Cycle 2012, 11, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.D.; Marchan, R.; Lesjak, M.S.; Lambert, J.; Hergenroeder, R.; Ellis, J.K.; Lau, C.-H.; Keun, H.C.; Schmitz, G.; Schiller, J.; et al. Choline-releasing glycerophosphodiesterase EDI3 drives tumour cell migration and metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 8155–8160. [Google Scholar] [CrossRef] [PubMed]
- Lesjak, M.S.; Marchan, R.; Stewart, J.D.; Rempel, E.; Rahnenführer, J.; Hengstler, J.G. EDI3 links choline metabolism to integrin expression, cell adhesion and spreading. Cell Adhes. Migr. 2014, 8, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Shinderman-Manan, E.; Cohen, K.; Weingarten, C.; Nabriski, D.; Twito, O.; Baraf, L.; Hercbergs, A.; Davis, P.J.; Werner, H.; Ellis, M.; et al. The thyroid hormone-αvβ3 integrin axis in ovarian cancer: Regulation of gene transcription and MAPK-dependent proliferation. Oncogene 2016, 35, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Ross, E.; Ata, R.; Thavarajah, T.; Medvedev, S.; Bowden, P.; Marshall, J.G.; Antonescu, C.N. AMP-activated protein kinase regulates the cell surface proteome and integrin membrane traffic. PLoS ONE 2015, 10, e0128013. [Google Scholar] [CrossRef] [PubMed]
- Cowden Dahl, K.D.; Robertson, S.E.; Weaver, V.M.; Simon, M.C. Hypoxia-inducible factor regulates αvβ3 integrin cell surface expression. Mol. Biol. Cell 2005, 16, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Rainero, E.; Howe, J.D.; Caswell, P.T.; Jamieson, N.B.; Anderson, K.; Critchley, D.R.; Machesky, L.; Norman, J.C. Ligand-occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 2015, 10, 398–413. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.; Karantza-Wadsworth, V.; White, E. Role of autophagy in cancer. Nat. Rev. Cancer 2007, 7, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [PubMed]
- Tuloup-Minguez, V.; Hamaï, A.; Greffard, A.; Nicolas, V.; Codogno, P.; Botti, J. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle 2013, 12, 3317–3328. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-O.; Shin, S.; Mercurio, A.M. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the α6β4 integrin. Cancer Res. 2005, 65, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-J.; Seo, S.-M.; Kim, E.J.; Lee, Y.-J.; Ko, Y.G.; Ha, J.; Lee, M. Berberine inhibits human colon cancer cell migration via AMP-activated protein kinase-mediated downregulation of integrin β1 signaling. Biochem. Biophys. Res. Commun. 2012, 426, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Draheim, K.M.; Chen, H.-B.; Tao, Q.; Moore, N.; Roche, M.; Lyle, S. ARRDC3 suppresses breast cancer progression by negatively regulating integrin β4. Oncogene 2010, 29, 5032–5047. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Emilsson, V.; Schadt, E.E.; Chutkow, W.A.; Lee, S.; Marsili, A.; Zhang, Y.; Dobrin, R.; Cohen, D.E.; Larsen, P.R.; et al. The arrestin domain-containing 3 protein regulates body mass and energy expenditure. Cell Metab. 2011, 14, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E. On the origins of arrestin and rhodopsin. BMC Evol. Biol. 2008, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Patwari, P.; Lee, R.T. An expanded family of arrestins regulate metabolism. Trends Endocrinol. Metab. 2012, 23, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Arboleda, M.J.; Lyons, J.F.; Kabbinavar, F.F.; Bray, M.R.; Snow, B.E.; Ayala, R.; Danino, M.; Karlan, B.Y.; Slamon, D.J. Overexpression of AKT2/protein kinase Bβ leads to up-regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003, 63, 196–206. [Google Scholar] [PubMed]
- Kitsiou, P.V.; Tzinia, A.K.; Stetler-Stevenson, W.G.; Michael, A.F.; Fan, W.-W.; Zhou, B.; Tsilibary, E.C. Glucose-induced changes in integrins and matrix-related functions in cultured human glomerular epithelial cells. Am. J. Physiol. Ren. Physiol. 2003, 284, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Karamessinis, P.; Tzinia, A.; Kitsiou, P.; Stetler-Stevenson, W.; Michael, A.F.; Fan, W.W.; Zhou, B.; Margaritis, L.H.; Tsilibary, E.C. Proximal tubular epithelial cell integrins respond to high glucose by altered cell-matrix interactions and differentially regulate matrixin expression. Lab. Investig. 2002, 82, 1081–1093. [Google Scholar] [CrossRef] [PubMed]
- Bellis, S.L. Variant glycosylation: An underappreciated regulatory mechanism for β1 integrins. Biochim. Biophys. Acta Biomembr. 2004, 1663, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, F.M.; Seales, E.C.; Clem, W.C.; Hennessy, K.M.; Zhuo, Y.; Bellis, S.L. Tumour cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp. Cell Res. 2008, 314, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.E.; Lityńska, A.; Vereecken, P. Cell migration—The role of integrin glycosylation. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Hang, Q.; Fukuda, T.; Isaji, T. Integrin α5β1 and its N-glycosylation. Glycoscience 2014. [Google Scholar] [CrossRef]
- Guo, H.-B.; Lee, I.; Kamar, M.; Akiyama, S.K.; Pierce, M. Aberrant N-glycosylation of β1 integrin causes reduced α5β1 integrin clustering and stimulates cell migration. Cancer Res. 2002, 62, 6837–6845. [Google Scholar] [PubMed]
- Nabi, I.R.; Shankar, J.; Dennis, J.W. The galectin lattice at a glance. J. Cell Sci. 2015, 128, 2213–2219. [Google Scholar] [CrossRef] [PubMed]
- Lagana, A.; Goetz, J.G.; Cheung, P.; Raz, A.; Dennis, J.W.; Nabi, I.R. Galectin binding to Mgat5-modified N-glycans regulates fibronectin matrix remodeling in tumour cells. Mol. Cell. Biol. 2006, 26, 3181–3193. [Google Scholar] [CrossRef] [PubMed]
- Fortuna-Costa, A.; Gomes, A.M.; Kozlowski, E.O.; Stelling, M.P.; Pavão, M.S.G. Extracellular galectin-3 in tumour progression and metastasis. Front. Oncol. 2014, 4, 138. [Google Scholar] [CrossRef] [PubMed]
- Shankar, J.; Boscher, C.; Nabi, I.R. Caveolin-1, galectin-3 and lipid raft domains in cancer cell signaling. Essays Biochem. 2015, 57, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Sasai, K.; Ikeda, Y.; Fujii, T.; Tsuda, T.; Taniguchi, N. UDP-GlcNAc concentration is an important factor in the biosynthesis of β1,6-branched oligosaccharides: Regulation based on the kinetic properties of N-acetylglucosaminyltransferase V. Glycobiology 2002, 12, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.S.; Partridge, E.A.; Grigorian, A.; Silvescu, C.I.; Reinhold, V.N.; Demetriou, M.; Dennis, J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007, 129, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Shirato, K.; Nakajima, K.; Korekane, H.; Takamatsu, S.; Gao, C.; Angata, T.; Ohtsubo, K.; Taniguchi, N. Hypoxic regulation of glycosylation via the N-acetylglucosamine cycle. J. Clin. Biochem. Nutr. 2011, 48, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.B.; Peixoto, J.; Soares, P.; Maximo, V.; Carvalho, S.; Pinho, S.S.; Vieira, A.F.; Paredes, J.; Rego, A.C.; Ferreira, I.L.; et al. OXPHOS dysfunction regulates integrin-β1 modifications and enhances cell motility and migration. Hum. Mol. Genet. 2015, 24, 1977–1990. [Google Scholar] [CrossRef] [PubMed]
- Büll, C.; Stoel, M.A.; den Brok, M.H.; Adema, G.J. Sialic acids sweeten a tumour’s life. Cancer Res. 2014, 74, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Yogeeswaran, G.; Salk, P. Metastatic potential is positively correlated with cell surface sialylation of cultured murine tumour cell lines. Science 1981, 212, 1514–1516. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Kemmner, W.; Grigull, S.; Schlag, P.M. Cell surface α2,6-sialylation affects adhesion of breast carcinoma cells. Exp. Cell Res. 2002, 276, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Pocheć, E.; Lityńska, A.; Amoresano, A.; Casbarra, A. Glycosylation profile of integrin α3β1 changes with melanoma progression. Biochim. Biophys. Acta Mol. Cell Res. 2003, 1643, 113–123. [Google Scholar] [CrossRef]
- Seales, E.C.; Jurado, G.A.; Brunson, B.A.; Wakefield, J.K.; Frost, A.R.; Bellis, S.L. Hypersialylation of β1 integrins, observed in colon adenocarcinoma, may contribute to cancer progression by up-regulating cell motility. Cancer Res. 2005, 65, 4645–4652. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Fan, J.; Liu, L.; Zhang, L.; Wang, S.; Zhang, J. Caveolin-1 up-regulates integrin α2,6-sialylation to promote integrin α5β1-dependent hepatocarcinoma cell adhesion. FEBS Lett. 2013, 587, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, L.; Shen, S.; Wu, S.; Burdick, M.M. Effect of α2,6 sialylation on integrin-mediated adhesion of breast cancer cells to fibronectin and collagen IV. Life Sci. 2016, 149, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.S.C.; Wati, L.; Nissom, P.M.; Feng, H.T.; Lee, M.M.; Yap, M.G. An investigation of intracellular glycosylation activities in CHO cells: Effects of nucleotide sugar precursor feeding. Biotechnol. Bioeng. 2010, 107, 321–336. [Google Scholar] [CrossRef] [PubMed]
- Almaraz, R.T.; Tian, Y.; Bhattarcharya, R.; Tan, E.; Chen, S.-H.; Dallas, M.R.; Chen, L.; Zhang, Z.; Zhang, H.; Konstantopoulos, K.; et al. Metabolic flux increases glycoprotein sialylation: Implications for cell adhesion and cancer metastasis. Mol. Cell. Proteom. 2012, 11, M112.017558. [Google Scholar] [CrossRef] [PubMed]
- Kucharzewska, P.; Christianson, H.C.; Belting, M. Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS ONE 2015, 10, e0116740. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gucek, M.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA 2008, 105, 13793–13798. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 80–95. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.D.; Hart, G.W. AMP-activated protein kinase and p38 MAPK activate O-GlcNAcylation of neuronal proteins during glucose deprivation. J. Biol. Chem. 2008, 283, 13009–13020. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Hoessli, D.C.; Walker-Nasir, E.; Choudhary, M.I.; Rafik, S.M.; Shakoori, A.R.; Nasir-ud-Din. Phosphorylation and glycosylation interplay: Protein modifications at hydroxy amino acids and prediction of signaling functions of the human β3 integrin family. J. Cell. Biochem. 2006, 99, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Kato, H.; Watanabe, T.; Min, K.-D.; Yamazaki, S.; Asano, Y.; Seguchi, O.; Higo, S.; Shintani, Y.; Asanuma, H.; et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat. Cell Biol. 2010, 12, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Tsukamoto, O.; Nakano, A.; Kato, H.; Kioka, H.; Higo, S.; Yamazaki, S.; Shintani, Y.; Matsuoka, K.; Liao, Y.; et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat. Commun. 2015, 6, 6137. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro-Smith, L.; Nagalingam, A.; Zhong, D.; Zhou, W.; Saxena, N.K.; Sharma, D. LKB1 is required for adiponectin-mediated modulation of AMPK-S6K axis and inhibition of migration and invasion of breast cancer cells. Oncogene 2009, 28, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Friedrich, D.; Chavakis, E.; Böhm, M.; Friedrich, E.B. Effect of hypoxia on integrin-mediated adhesion of endothelial progenitor cells. J. Cell. Mol. Med. 2012, 16, 2387–2393. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.; Kim, H.; Nam, K.; Shin, I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep. 2016, in press. [Google Scholar]
- Hahn, S.S.; Tang, Q.; Zheng, F.; Zhao, S.; Wu, J.; Chen, J. Repression of integrin-linked kinase by antidiabetes drugs through cross-talk of PPARγ- and AMPKα-dependent signaling: Role of AP-2α and Sp1. Cell Signal. 2014, 26, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Zhao, S.; Wu, J.; Zheng, F.; Yang, L.; Wu, J.; Chen, J. Inhibition of integrin-linked kinase expression by emodin through crosstalk of AMPKα and ERK1/2 signaling and reciprocal interplay of Sp1 and c-Jun. Cell Signal. 2015, 27, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Corley, K.M.; Taylor, C.J.; Lilly, B. Hypoxia-inducible factor 1α modulates adhesion, migration, and FAK phosphorylation in vascular smooth muscle cells. J. Cell. Biochem. 2005, 96, 971–985. [Google Scholar] [CrossRef] [PubMed]
- Kline, E.R.; Shupe, J.; Gilbert-Ross, M.; Zhou, W.; Marcus, A.I. LKB1 represses focal adhesion kinase (FAK) signaling via a FAK-LKB1 complex to regulate FAK site maturation and directional persistence. J. Biol. Chem. 2013, 288, 17663–17674. [Google Scholar] [CrossRef] [PubMed]
- Rantala, J.K.; Pouwels, J.; Pellinen, T.; Veltel, S.; Laasola, P.; Mattila, E.; Potter, C.S.; Duffy, T.; Sundberg, J.P.; Kallioniemi, O.; et al. SHARPIN is an endogenous inhibitor of β1-integrin activation. Nat. Cell Biol. 2011, 13, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.L.; Leprivier, G.; Robertson, M.D.; Chow, C.; Martin, M.J.; Laderoute, K.R.; Davicioni, E.; Triche, T.J.; Sorensen, P.H. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012, 19, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Hindupur, S.K.; Balaji, S.A.; Saxena, M.; Pandey, S.; Sravan, G.S.; Heda, N.; Kumar, M.V.; Mukherjee, G.; Dey, D.; Rangarajan, A. Identification of a novel AMPK-PEA15 axis in the anoikis-resistant growth of mammary cells. Breast Cancer Res. 2014, 16, 420. [Google Scholar] [CrossRef] [PubMed]
- Sundararaman, A.; Amirtham, U.; Rangarajan, A. Calcium-oxidant signaling network regulates AMP-activated Protein Kinase (AMPK) Activation upon Matrix Deprivation. J. Biol. Chem. 2016, 291, 14410–14429. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Stock, C.; Gassner, B.; Hauck, C.R.; Arnold, H.; Mally, S.; Mally, S.; Eble, J.A.; Dieterich, P.; Schwab, A. Migration of human melanoma cells depends on extracellular pH and Na+/H+ exchange. J. Physiol. 2005, 567, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Paradise, R.K.; Lauffenburger, D.A.; van Vliet, K.J. Acidic extracellular pH promotes activation of integrin αvβ3. PLoS ONE 2011, 6, e15746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, N.K.; Gaffney, E.A.; Gatenby, R.A.; Maini, P.K. Tumour-stromal interactions in acid-mediated invasion: A mathematical model. J. Theor. Biol. 2010, 267, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Wu, C. ILK: A pseudokinase in the center stage of cell-matrix adhesion and signaling. Curr. Opin. Cell Biol. 2012, 24, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Nollet, E.A.; Miranti, C.K. Integrin and adhesion regulation of autophagy and mitophagy. autophagy—A double-edged sword—Cell survival or death? InTech 2013. [Google Scholar] [CrossRef]
- Serrano, I.; McDonald, P.C.; Lock, F.E.; Dedhar, S. Role of the integrin-linked kinase (ILK)/Rictor complex in TGFβ-1-induced epithelial–mesenchymal transition (EMT). Oncogene 2013, 32, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Shaul, Y.D.; Freinkman, E.; Comb, W.C.; Cantor, J.R.; Tam, W.L.; Thiru, P.; Kim, D.; Kanarek, N.; Pacold, M.E.; Chen, W.W.; et al. Dihydropyrimidine accumulation is required for the epithelial-mesenchymal transition. Cell 2014, 158, 1094–1109. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.-L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Serrano, I.; McDonald, P.C.; Lock, F.; Muller, W.J.; Dedhar, S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 2013, 4, 2976. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the tumour suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.; Werb, Z. Integrins engage mitochondrial function for signal transduction by a mechanism dependent on Rho GTPases. J. Cell Biol. 2002, 158, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Hou, Y.; Yuan, J.; Tang, S.; Zhang, H.; Zhu, Q.; Du, Y.E.; Zhou, M.; Wen, S.; Xu, L.; et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget 2015, 6, 25755–25769. [Google Scholar] [CrossRef] [PubMed]
- Rintoul, R.C. Cross-linking CD98 promotes integrin-like signaling and anchorage-independent growth. Mol. Biol. Cell 2002, 13, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Fenczik, C.A.; Sethi, T.; Ramos, J.W.; Hughes, P.E.; Ginsberg, M.H. Complementation of dominant suppression implicates CD98 in integrin activation. Nature 1997, 390, 81–85. [Google Scholar] [PubMed]
- Zent, R.; Fenczik, C.A.; Calderwood, D.A.; Liu, S.; Dellos, M.; Ginsberg, M.H. Class- and splice variant-specific association of CD98 with integrin β cytoplasmic domains. J. Biol. Chem. 2000, 275, 5059–5064. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Bulus, N.; Fonseca-Siesser, P.M.; Chen, D.; Hanks, S.K.; Pozzi, A.; Zent, R. CD98 modulates integrin β1 function in polarized epithelial cells. J. Cell Sci. 2005, 118, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.M.; Ginsberg, M.H. CD98 at the crossroads of adaptive immunity and cancer. J. Cell Sci. 2012, 125, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-M.; Hahn, J.-H. CD98 activation increases surface expression and clustering of β1 integrins in MCF-7 cells through FAK/Src- and cytoskeleton-independent mechanisms. Exp. Mol. Med. 2008, 40, 261. [Google Scholar] [CrossRef] [PubMed]
- Fenczik, C.A.; Zent, R.; Dellos, M.; Calderwood, D.A.; Satriano, J.; Kelly, C.; Ginsberg, M.H. Distinct domains of CD98hc regulate integrins and amino acid transport. J. Biol. Chem. 2001, 276, 8746–8752. [Google Scholar] [CrossRef] [PubMed]
- Feral, C.C.; Nishiya, N.; Fenczik, C.A.; Stuhlmann, H.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) mediates integrin signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.M.; Castorino, J.J.; Philp, N.J. Interaction of monocarboxylate transporter 4 with β1-integrin and its role in cell migration. Am. J. Physiol. Cell Physiol. 2009, 296, C414–C421. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, C.; Longatto-Filho, A.; Azevedo-Silva, J.; Casal, M.; Schmitt, F.C.; Baltazar, F. Role of monocarboxylate transporters in human cancers: State of the art. J. Bioenerg. Biomembr. 2012, 44, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Hagner, N.; Joerger, M. Cancer chemotherapy: Targeting folic acid synthesis. Cancer Manag. Res. 2010, 2, 293–301. [Google Scholar] [PubMed]
- Maiuthed, A.; Chanvorachote, P. Cisplatin at sub-toxic levels mediates integrin switch in lung cancer cells. Anticancer Res. 2014, 34, 7111–7117. [Google Scholar] [PubMed]
- Tsaur, I.; Makarević, J.; Juengel, E.; Gasser, M.; Waaga-Gasser, A.-M.; Kurosch, M.; Reiter, M.; Wedel, S.; Bartsch, G.; Haferkamp, A.; et al. Resistance to the mTOR-inhibitor RAD001 elevates integrin α2- and β1-triggered motility, migration and invasion of prostate cancer cells. Br. J. Cancer 2012, 107, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Ambriovic-Ristov, A.; Osmak, M. Integrin-mediated drug resistance. Curr. Signal. Transduct. Ther. 2006, 1, 227–237. [Google Scholar] [CrossRef]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; Pusztai, L.; Swanton, C. Cancer heterogeneity: Implications for targeted therapeutics. Br. J. Cancer 2013, 108, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Polyak, K. Tumour heterogeneity: Causes and consequences. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; et al. Molecular definition of breast tumour heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Almendro, V.; Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 2012, 12, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumour heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Soeda, A.; Hara, A.; Kunisada, T.; Yoshimura, S.; Iwama, T.; Park, D.M. The evidence of glioblastoma heterogeneity. Sci. Rep. 2015, 5, 7979. [Google Scholar] [CrossRef] [PubMed]
- Zenobi, R. Single-cell metabolomics: Analytical and biological perspectives. Science 2013, 342, 1243259. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.; Zheng, B.; Hsu, C.-H.; Sasaki, A.T.; Cantley, L.C. A fluorescent reporter of AMPK activity and cellular energy stress. Cell Metab. 2011, 13, 476–486. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumours. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ata, R.; Antonescu, C.N. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. Int. J. Mol. Sci. 2017, 18, 189. https://doi.org/10.3390/ijms18010189
Ata R, Antonescu CN. Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. International Journal of Molecular Sciences. 2017; 18(1):189. https://doi.org/10.3390/ijms18010189
Chicago/Turabian StyleAta, Rehman, and Costin N. Antonescu. 2017. "Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer" International Journal of Molecular Sciences 18, no. 1: 189. https://doi.org/10.3390/ijms18010189
APA StyleAta, R., & Antonescu, C. N. (2017). Integrins and Cell Metabolism: An Intimate Relationship Impacting Cancer. International Journal of Molecular Sciences, 18(1), 189. https://doi.org/10.3390/ijms18010189