
Modulation of the Unfolded Protein Response by Tauroursodeoxycholic Acid Counteracts Apoptotic Cell Death and Fibrosis in a Mouse Model for Secondary Biliary Liver Fibrosis



 ,
,
Abstract
:1. Introduction
2. Results
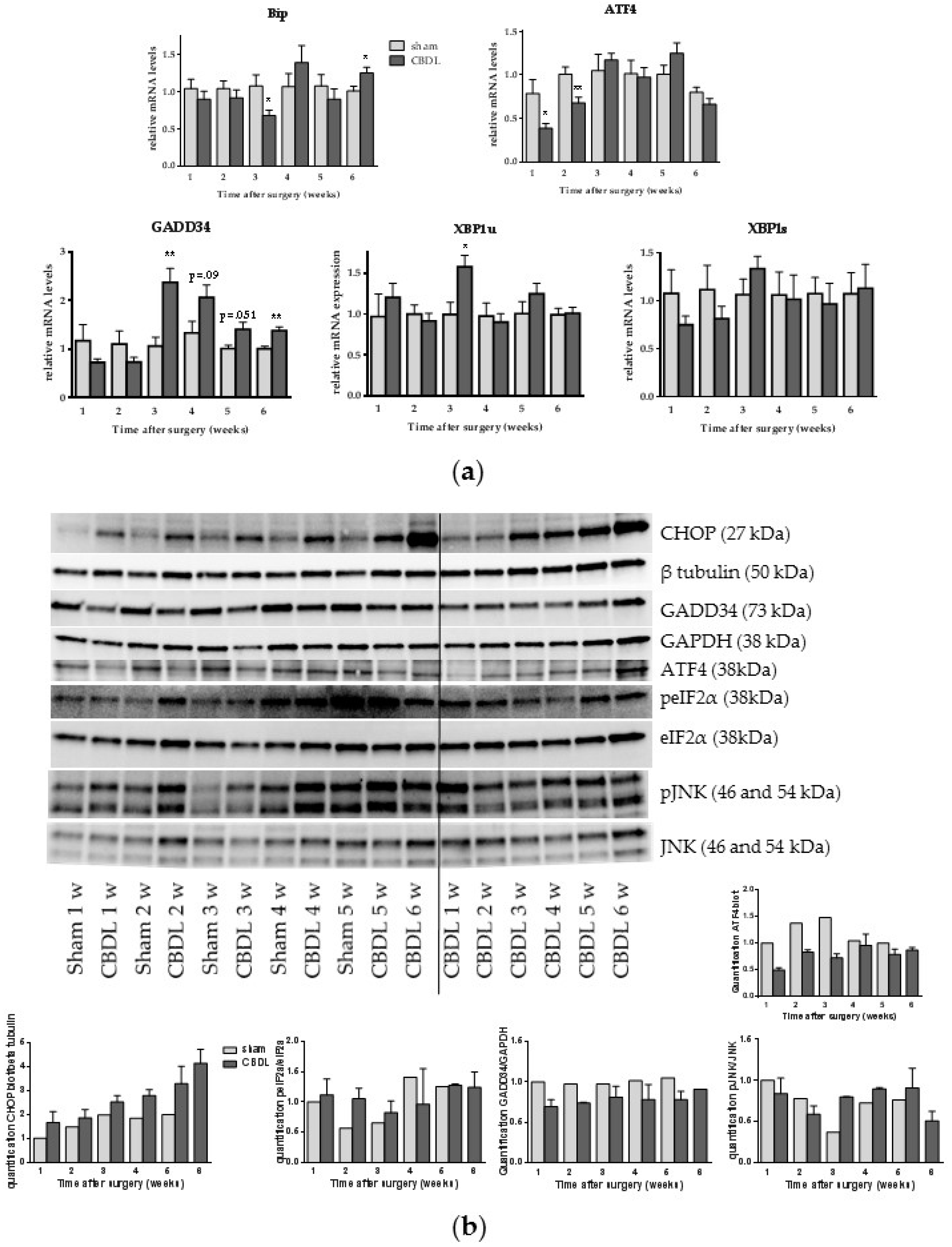
2.1. Common Bile Duct Ligation Activates the Hepatic Unfolded Protein Response
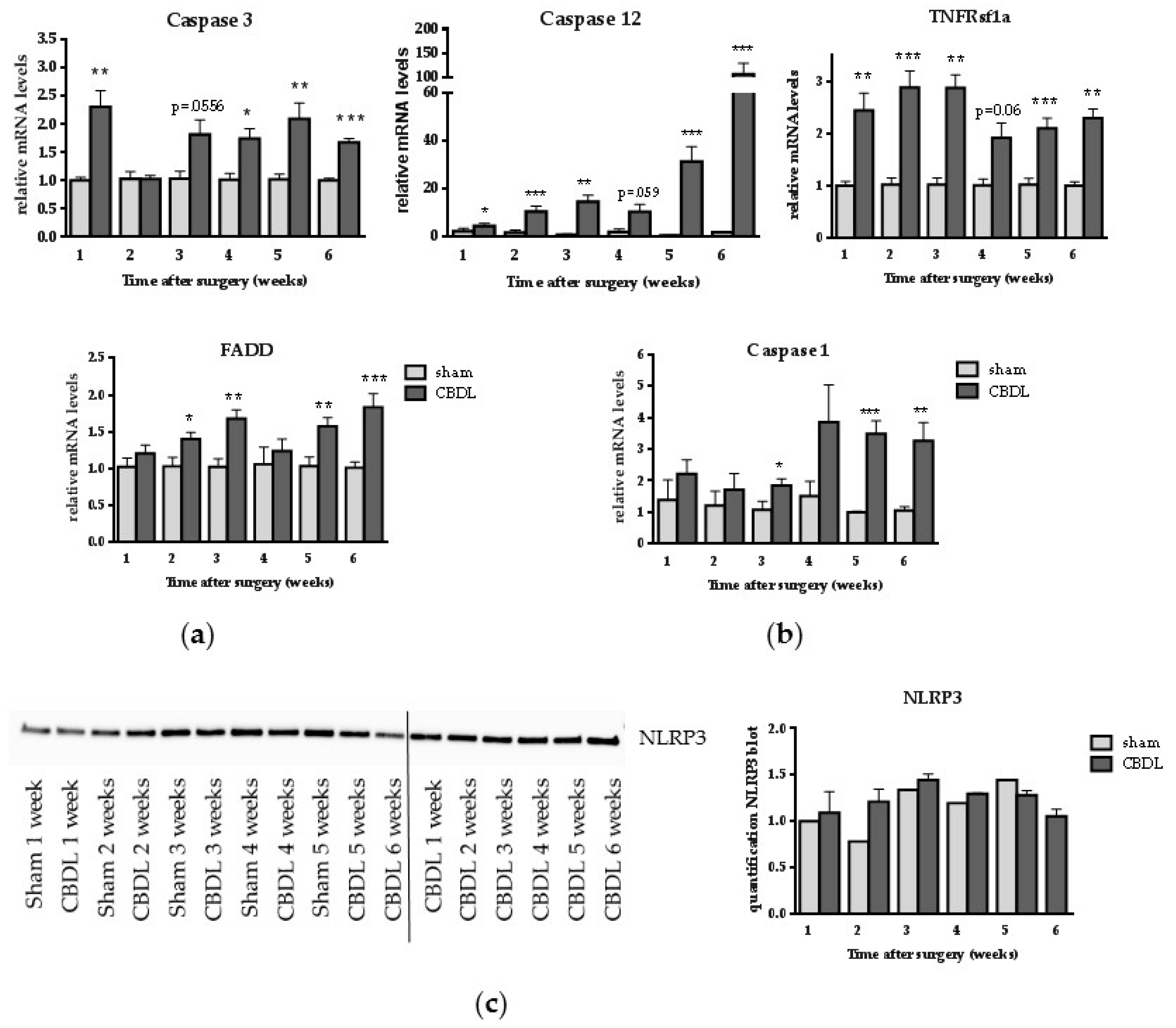
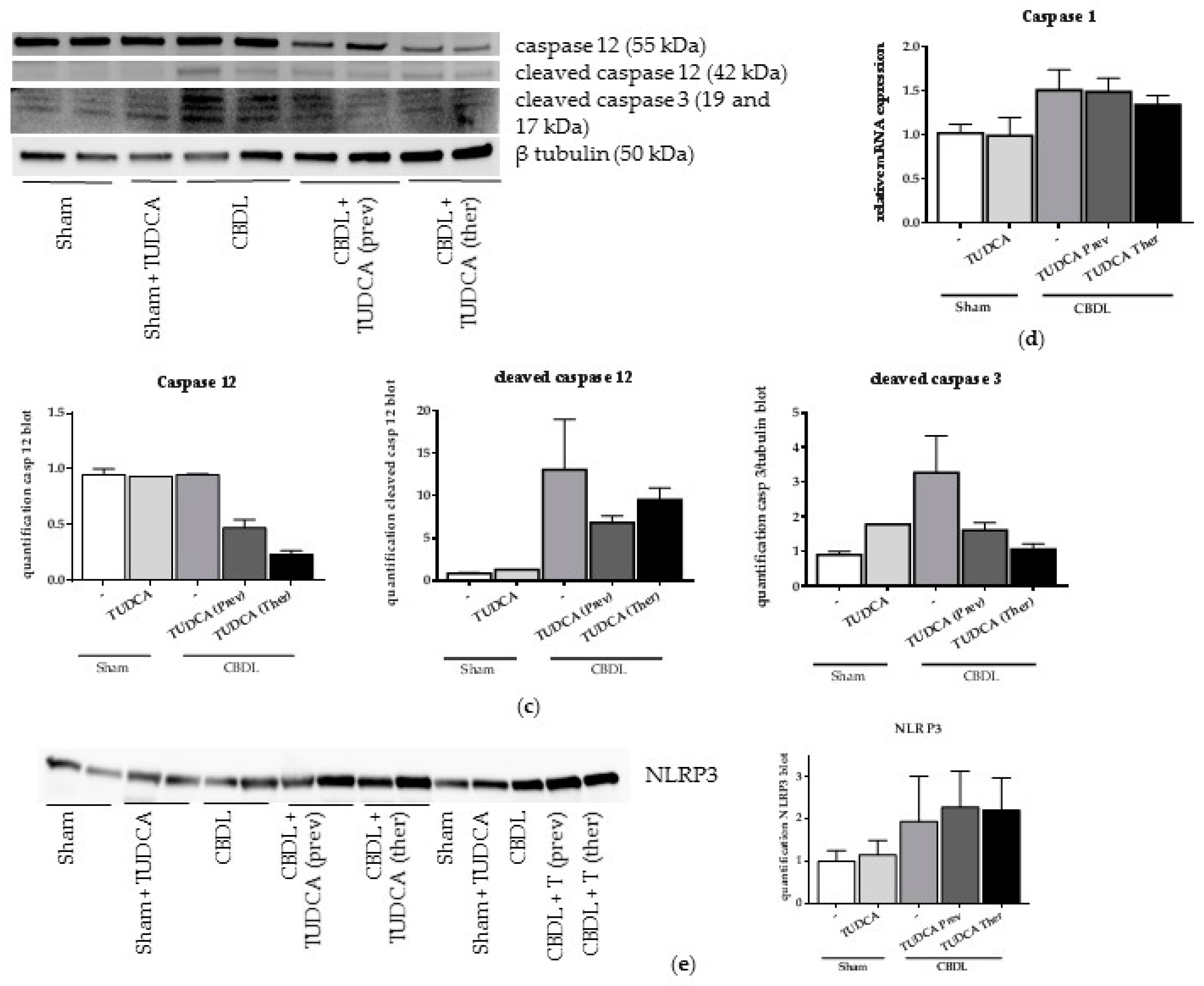
2.2. CBDL Induces a Pro-Apoptotic Response
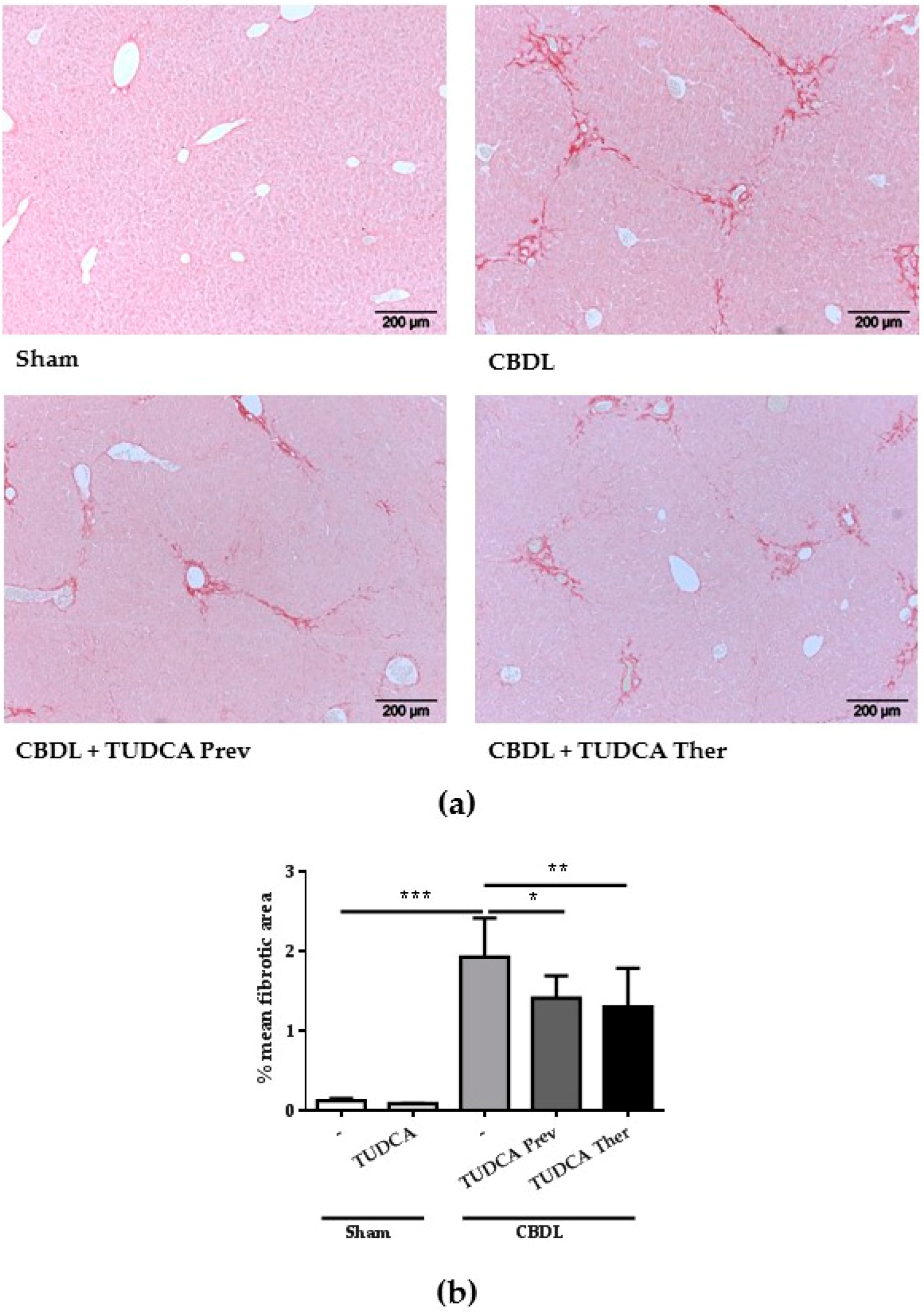
2.3. Treatment with TUDCA Reduces the Degree of Liver Fibrosis
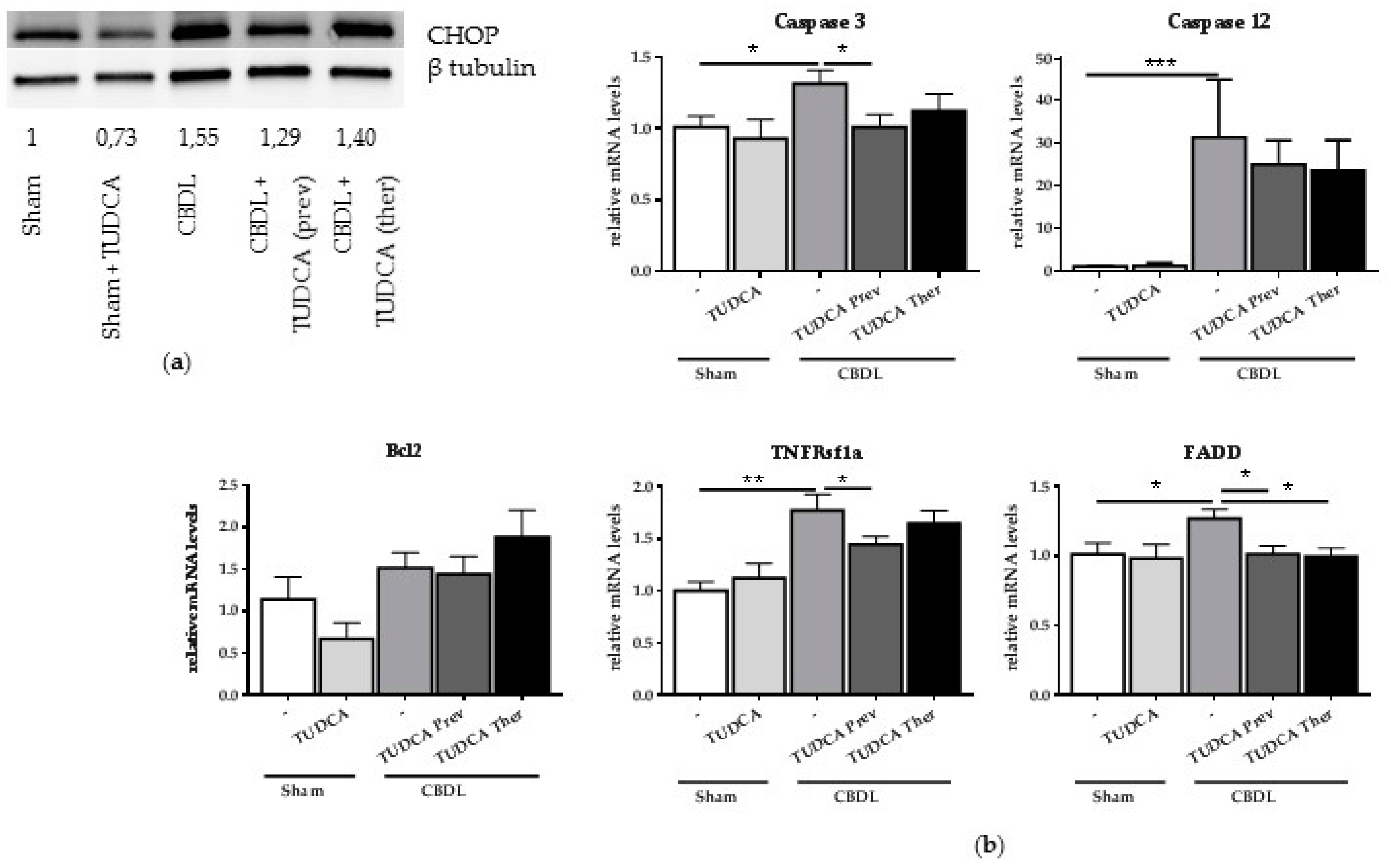
2.4. Treatment with TUDCA Reduces CBDL-Induced CHOP and Pro-Apoptotic Markers
3. Discussion
3.1. CBDL Induces Hepatic Unfolded Protein and Pro-Apoptotic Response
3.2. Treatment with TUDCA Reduces Liver Damage and Liver Fibrosis through Modulation of CHOP and Its Pro-Apoptotic Response
4. Materials and Methods
4.1. Animals
4.2. Induction of Secondary Biliary Fibrosis
4.3. Kinetics of Hepatic UPR and Cell Death in Cholestasis-Induced Liver Fibrosis
4.4. Preventive and Therapeutic Study with Tauroursodeoxycholic Acid
4.5. Evaluation of Liver Fibrosis
4.6. qPCR
4.7. Western Blotting
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hofmann, A.F. Cholestatic liver disease: Pathophysiology and therapeutic options. Liver 2002, 22, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Hirschfield, G.M.; Heathcote, E.J.; Gershwin, M.E. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 2010, 139, 1481–1496. [Google Scholar] [CrossRef] [PubMed]
- Perez, M.J.; Briz, O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Schattenberg, J.M.; Zimmermann, T.; Worns, M.; Sprinzl, M.F.; Kreft, A.; Kohl, T.; Nagel, M.; Siebler, J.; Schulze Bergkamen, H.; He, Y.W.; et al. Ablation of c-flip in hepatocytes enhances death-receptor mediated apoptosis and toxic liver injury in vivo. J. Hepatol. 2011, 55, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Payne, C.M.; Bernstein, C.; Schneider, J.; Beard, S.E.; Crowley, C.L. Activation of the promoters of genes associated with DNA damage, oxidative stress, ER stress and protein malfolding by the bile salt, deoxycholate. Toxicol. Lett. 1999, 108, 37–46. [Google Scholar] [CrossRef]
- Dai, B.H.; Geng, L.; Wang, Y.; Sui, C.J.; Xie, F.; Shen, R.X.; Shen, W.F.; Yang, J.M. MicroRNA-199a-5p protects hepatocytes from bile acid-induced sustained endoplasmic reticulum stress. Cell Death Dis. 2013, 4, e604. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, S.; Tsuji, M.; Morio, Y.; Oguchi, K. Involvement of endoplasmic reticulum in glycochenodeoxycholic acid-induced apoptosis in rat hepatocytes. Toxicol. Lett. 2006, 166, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Bochkis, I.M.; Rubins, N.E.; White, P.; Furth, E.E.; Friedman, J.R.; Kaestner, K.H. Hepatocyte-specific ablation of Foxa2 alters bile acid homeostasis and results in endoplasmic reticulum stress. Nat. Med. 2008, 14, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, N.; Hatano, E.; Taura, K.; Tada, M.; Kodama, Y.; Nitta, T.; Iwaisako, K.; Seo, S.; Nakajima, A.; Ikai, I.; et al. Chop deficiency attenuates cholestasis-induced liver fibrosis by reduction of hepatocyte injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G498–G505. [Google Scholar] [CrossRef] [PubMed]
- Uzi, D.; Barda, L.; Scaiewicz, V.; Mills, M.; Mueller, T.; Gonzalez-Rodriguez, A.; Valverde, A.M.; Iwawaki, T.; Nahmias, Y.; Xavier, R.; et al. Chop is a critical regulator of acetaminophen-induced hepatotoxicity. J. Hepatol. 2013, 59, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed]
- Benz, C.; Angermuller, S.; Otto, G.; Sauer, P.; Stremmel, W.; Stiehl, A. Effect of tauroursodeoxycholic acid on bile acid-induced apoptosis in primary human hepatocytes. Eur. J. Clin. Investig. 2000, 30, 203–209. [Google Scholar] [CrossRef]
- Falasca, L.; Tisone, G.; Palmieri, G.; Anselmo, A.; Di Paolo, D.; Baiocchi, L.; Torri, E.; Orlando, G.; Casciani, C.U.; Angelico, M. Protective role of tauroursodeoxycholate during harvesting and cold storage of human liver: A pilot study in transplant recipients. Transplantation 2001, 71, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Khaoustov, V.I.; Chung, C.C.; Sohn, J.; Krishnan, B.; Lewis, D.E.; Yoffe, B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology 2002, 36, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.; Moshage, H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, U.; Yilmaz, E.; Ozcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Gorgun, C.Z.; Hotamisligil, G.S. Chemical chaperones reduce er stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef] [PubMed]
- Pusl, T.; Vennegeerts, T.; Wimmer, R.; Denk, G.U.; Beuers, U.; Rust, C. Tauroursodeoxycholic acid reduces bile acid-induced apoptosis by modulation of AP-1. Biochem. Biophys. Res. Commun. 2008, 367, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Amaral, J.D.; Viana, R.J.; Ramalho, R.M.; Steer, C.J.; Rodrigues, C.M. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.Y.; Hong, S.H.; Lee, Y.J.; Chung, S.S.; Jung, H.S.; Park, S.G.; Park, K.S. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem. Biophys. Res. Commun. 2010, 397, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Ben Mosbah, I.; Alfany-Fernandez, I.; Martel, C.; Zaouali, M.A.; Bintanel-Morcillo, M.; Rimola, A.; Rodes, J.; Brenner, C.; Rosello-Catafau, J.; Peralta, C. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia-reperfusion. Cell Death Dis. 2010, 1, e52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, A.S.; Dewey, A.M.; Anderson, K.A.; Olivares, S.; Green, R.M. Reducing endoplasmic reticulum stress does not improve steatohepatitis in mice fed a methionine- and choline-deficient diet. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G54–G59. [Google Scholar] [CrossRef] [PubMed]
- Laukens, D.; Devisscher, L.; van den Bossche, L.; Hindryckx, P.; Vandenbroucke, R.E.; Vandewynckel, Y.P.; Cuvelier, C.; Brinkman, B.M.; Libert, C.; Vandenabeele, P.; et al. Tauroursodeoxycholic acid inhibits experimental colitis by preventing early intestinal epithelial cell death. Lab. Investig. 2014, 94, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.S.; Zimmermann, E.M.; Chuang, B.M.; Song, B.; Nwokoye, A.; Wilkinson, J.E.; Eaton, K.A.; Kaufman, R.J. The unfolded protein response and chemical chaperones reduce protein misfolding and colitis in mice. Gastroenterology 2013, 144, 989.e6–1000.e6. [Google Scholar] [CrossRef] [PubMed]
- Siddesha, J.M.; Nakada, E.M.; Mihavics, B.R.; Hoffman, S.M.; Rattu, G.K.; Chamberlain, N.; Cahoon, J.M.; Lahue, K.G.; Daphtary, N.; Aliyeva, M.; et al. Effect of a chemical chaperone, tauroursodeoxycholic acid, on HDM-induced allergic airway disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L1243–L1259. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Gershwin, M.E.; Poupon, R.; Kaplan, M.; Bergasa, N.V.; Heathcote, E.J. Primary biliary cirrhosis. Hepatology 2009, 50, 291–308. [Google Scholar] [CrossRef] [PubMed]
- Gani, A.R.; Uppala, J.K.; Ramaiah, K.V. Tauroursodeoxycholic acid prevents stress induced aggregation of proteins in vitro and promotes perk activation in HEPG2 cells. Arch. Biochem. Biophys. 2015, 568, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Yoshimura-Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J. Gastroenterol. 2015, 50, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.; Seki, E.; Osawa, Y.; Kodama, Y.; De Minicis, S.; Knowles, M.; Brenner, D.A. α-1 antitrypsin z protein (PiZ) increases hepatic fibrosis in a murine model of cholestasis. Hepatology 2007, 46, 1443–1452. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Li, Y.; Cheng, X.; Li, H. ER stress contributes to α-naphthyl isothiocyanate-induced liver injury with cholestasis in mice. Pathol. Res. Pract. 2016, 212, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Kaufman, R.J. From acute er stress to physiological roles of the unfolded protein response. Cell Death Differ. 2006, 13, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yao, W.; Xu, J.; Qiu, Y.; Cao, F.; Li, S.; Yang, S.; Yang, H.; Wu, Z.; Hou, Y. The anti-inflammatory effects of acetaminophen and N-acetylcysteine through suppression of the NLRP3 inflammasome pathway in LPS-challenged piglet mononuclear phagocytes. Innate Immun. 2015, 21, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Yun, K.J.; Nan, J.X.; Sohn, D.H.; Lee, B.H. Changes in expression and immunolocalization of protein associated with toxic bile salts-induced apoptosis in rat hepatocytes. Arch. Toxicol. 2003, 77, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Demirbilek, S.; Tas, E.; Gurunluoglu, K.; Akin, M.; Aksoy, R.T.; Emre, M.H.; Aydin, N.E.; Ay, S.; Ozatay, N. Fluvastatin reduced liver injury in rat model of extrahepatic cholestasis. Pediatr. Surg. Int. 2007, 23, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Li, T.W.; Ko, K.S.; Xia, M.; Lu, S.C. Switch from Mnt-Max to Myc-Max induces p53 and cyclin D1 expression and apoptosis during cholestasis in mouse and human hepatocytes. Hepatology 2009, 49, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Paumgartner, G.; Beuers, U. Ursodeoxycholic acid in cholestatic liver disease: Mechanisms of action and therapeutic use revisited. Hepatology 2002, 36, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Z.; Zhao, S.; Xiang, R. Chemical chaperones reduce ER stress and adipose tissue inflammation in high fat diet-induced mouse model of obesity. Sci. Rep. 2016, 6, 27486. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.L.; Zhao, L.; Li, L.; Li, A.H.; Ye, J.; Yang, L.; Xu, K.S.; Hou, X.H. Efficacy and safety of tauroursodeoxycholic acid in the treatment of liver cirrhosis: A double-blind randomized controlled trial. J. Huazhong Univ. Sci. Technol. 2013, 33, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, K.; Kinbara, S.; Miyazawa, N.; Takeuchi, Y.; Hirabayashi, N.; Kasai, H.; Araki, T. Effect of sodium tauroursodeoxycholate (UR-906) on liver dysfunction in bile duct-ligated rats. Eur. J. Pharmacol. 1997, 333, 207–213. [Google Scholar] [CrossRef]
- Hatipoglu, A.R.; Oguz, S.; Gurcan, S.; Yalta, T.; Albayrak, D.; Erenoglu, C.; Sagiroglu, T.; Sezer, Y.A. Combined effects of tauroursodeoxycholic acid and glutamine on bacterial translocation in obstructive jaundiced rats. Balkan Med. J. 2013, 30, 362–368. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Number of Animals | ALT | AST |
|---|---|---|---|
| sham | 6 | 24.05 ± 5.60 | 101.20 ± 36.17 |
| sham + TUDCA | 7 | 27.23 ± 8.36 | 96.43 ± 36.84 |
| CBDL | 9 | 312.3 ± 85.26 *** | 505.3 ± 124 *** |
| CBDL + TUDCA preventive | 10 | 274.4 ± 68.75 | 480.0 ± 121.8 |
| CBDL + TUDCA therapeutic | 8 | 246.3 ± 52.54 p = 0.095 | 413.4 ± 94.49 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paridaens, A.; Raevens, S.; Devisscher, L.; Bogaerts, E.; Verhelst, X.; Hoorens, A.; Van Vlierberghe, H.; Van Grunsven, L.A.; Geerts, A.; Colle, I. Modulation of the Unfolded Protein Response by Tauroursodeoxycholic Acid Counteracts Apoptotic Cell Death and Fibrosis in a Mouse Model for Secondary Biliary Liver Fibrosis. Int. J. Mol. Sci. 2017, 18, 214. https://doi.org/10.3390/ijms18010214
Paridaens A, Raevens S, Devisscher L, Bogaerts E, Verhelst X, Hoorens A, Van Vlierberghe H, Van Grunsven LA, Geerts A, Colle I. Modulation of the Unfolded Protein Response by Tauroursodeoxycholic Acid Counteracts Apoptotic Cell Death and Fibrosis in a Mouse Model for Secondary Biliary Liver Fibrosis. International Journal of Molecular Sciences. 2017; 18(1):214. https://doi.org/10.3390/ijms18010214
Chicago/Turabian StyleParidaens, Annelies, Sarah Raevens, Lindsey Devisscher, Eliene Bogaerts, Xavier Verhelst, Anne Hoorens, Hans Van Vlierberghe, Leo A. Van Grunsven, Anja Geerts, and Isabelle Colle. 2017. "Modulation of the Unfolded Protein Response by Tauroursodeoxycholic Acid Counteracts Apoptotic Cell Death and Fibrosis in a Mouse Model for Secondary Biliary Liver Fibrosis" International Journal of Molecular Sciences 18, no. 1: 214. https://doi.org/10.3390/ijms18010214
APA StyleParidaens, A., Raevens, S., Devisscher, L., Bogaerts, E., Verhelst, X., Hoorens, A., Van Vlierberghe, H., Van Grunsven, L. A., Geerts, A., & Colle, I. (2017). Modulation of the Unfolded Protein Response by Tauroursodeoxycholic Acid Counteracts Apoptotic Cell Death and Fibrosis in a Mouse Model for Secondary Biliary Liver Fibrosis. International Journal of Molecular Sciences, 18(1), 214. https://doi.org/10.3390/ijms18010214