
α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress

Abstract
:
1. Introduction
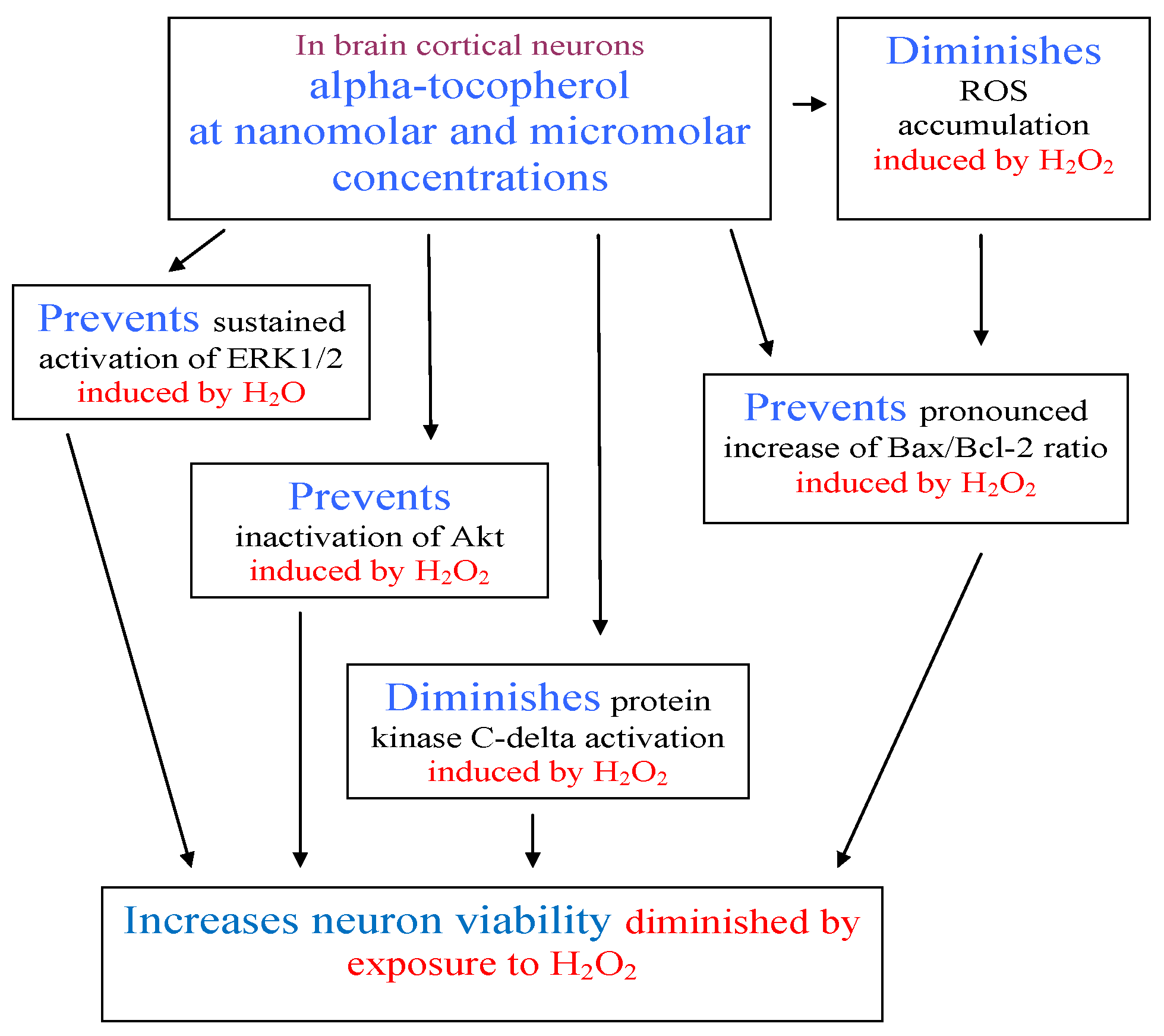
2. Results and Discussion
2.1. α-T and the Viability and Function of Nerve Cells
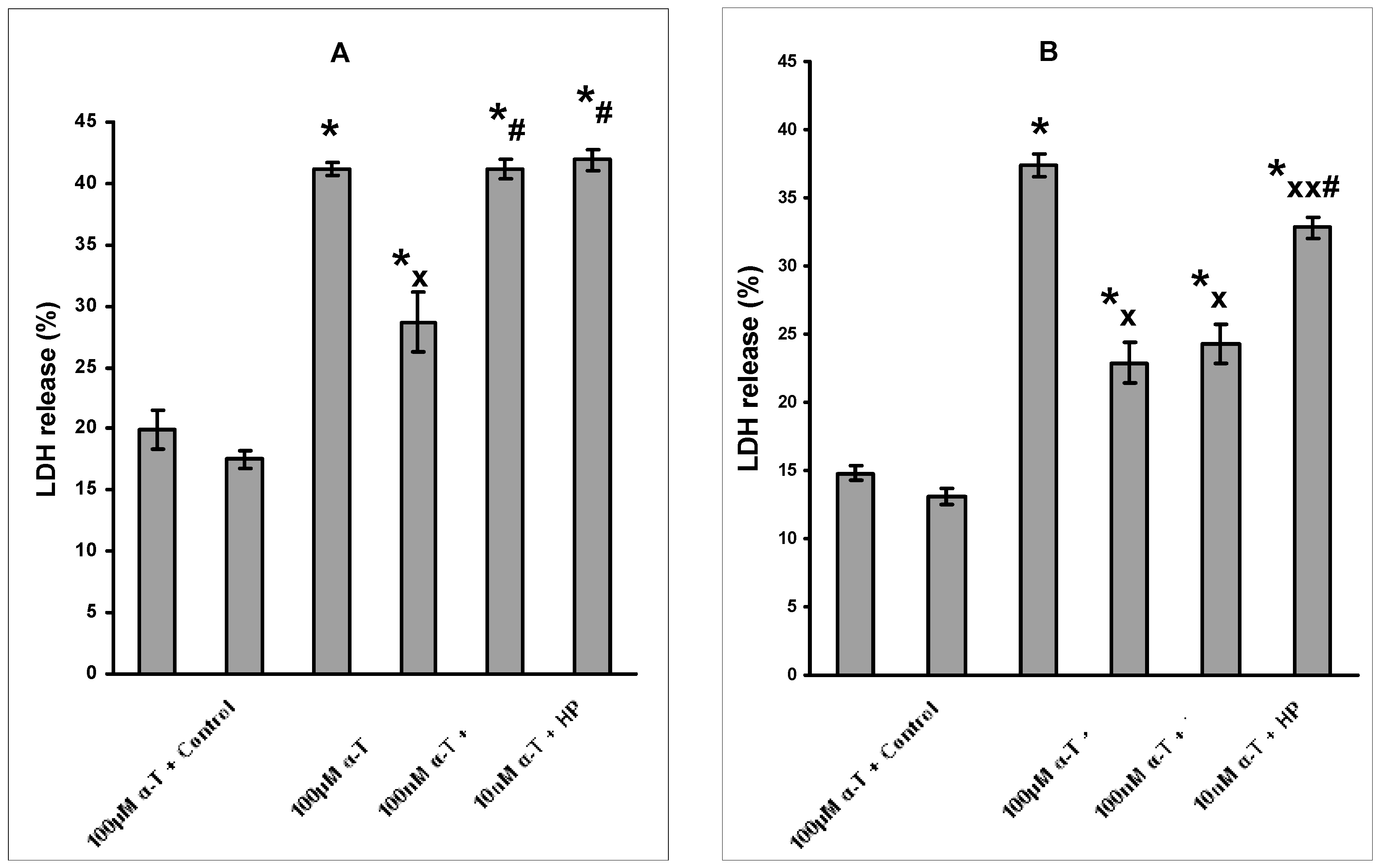
2.2. The Dosage and Duration of Treatment with α-T Govern Its Neuroprotective Effect
2.3. The Protective Effect of Preincubation with α-T for 18 h against H2O2-Induced Death of Brain Cortical Neurons Is Diminished or Abolished in the Presence of Inhibitors of PI 3-Kinase, MEK1/2 and PKCδ
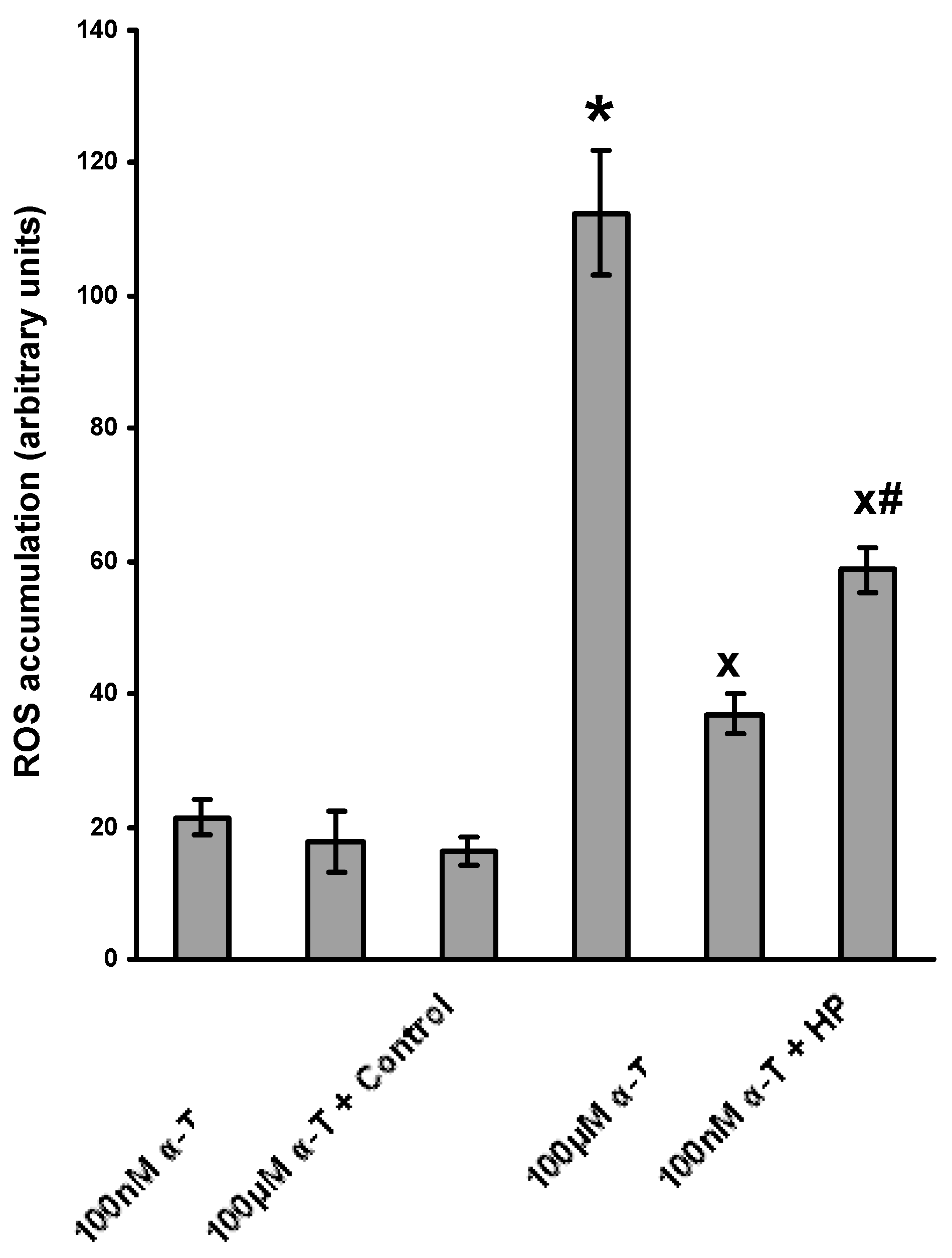
2.4. α-T at Micromolar and Nanomolar Concentrations Diminishes the Accumulation of ROS Induced in Brain Cortical Neurons by H2O2
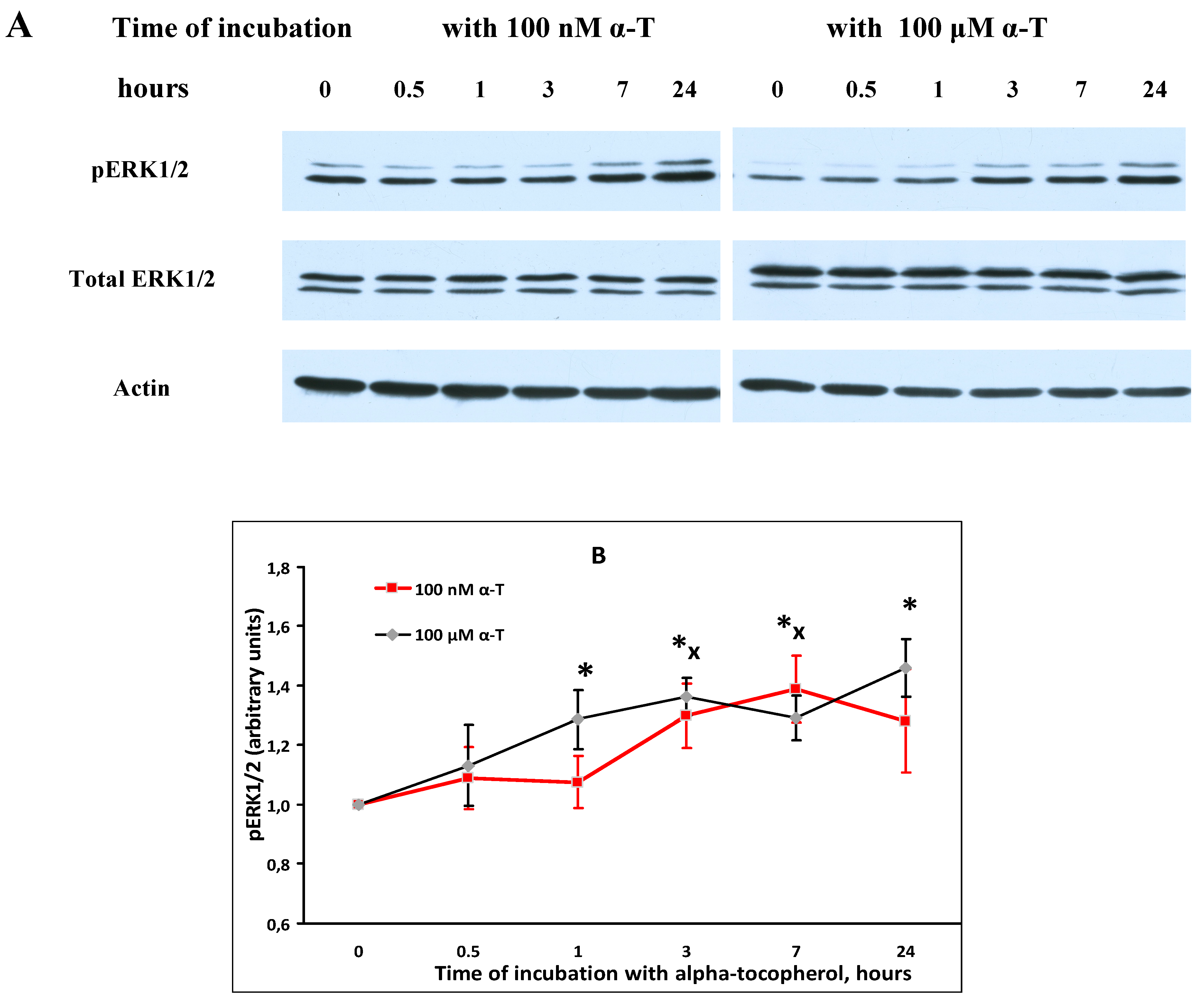
2.5. α-T Increases the Basal Level of pAkt and pERK1/2 in Brain Cortical Neurons, but Does Not Change Total Akt and ERK1/2 Levels
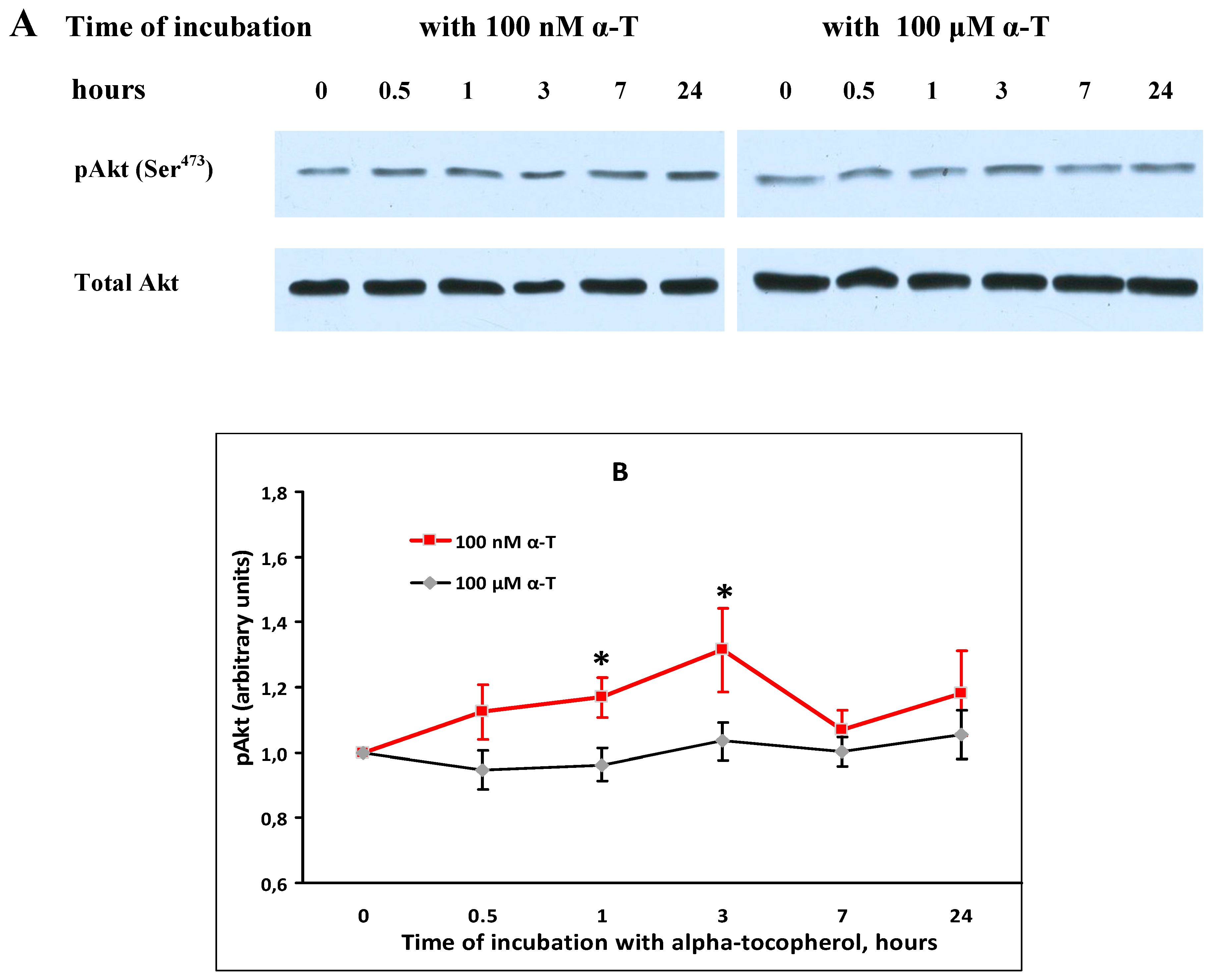
2.6. While α-T Prevents Akt Inactivation Initiated by H2O2 in Brain Cortical Neurons, the Effect of 100 nM and 100 μM α-T Is Similar
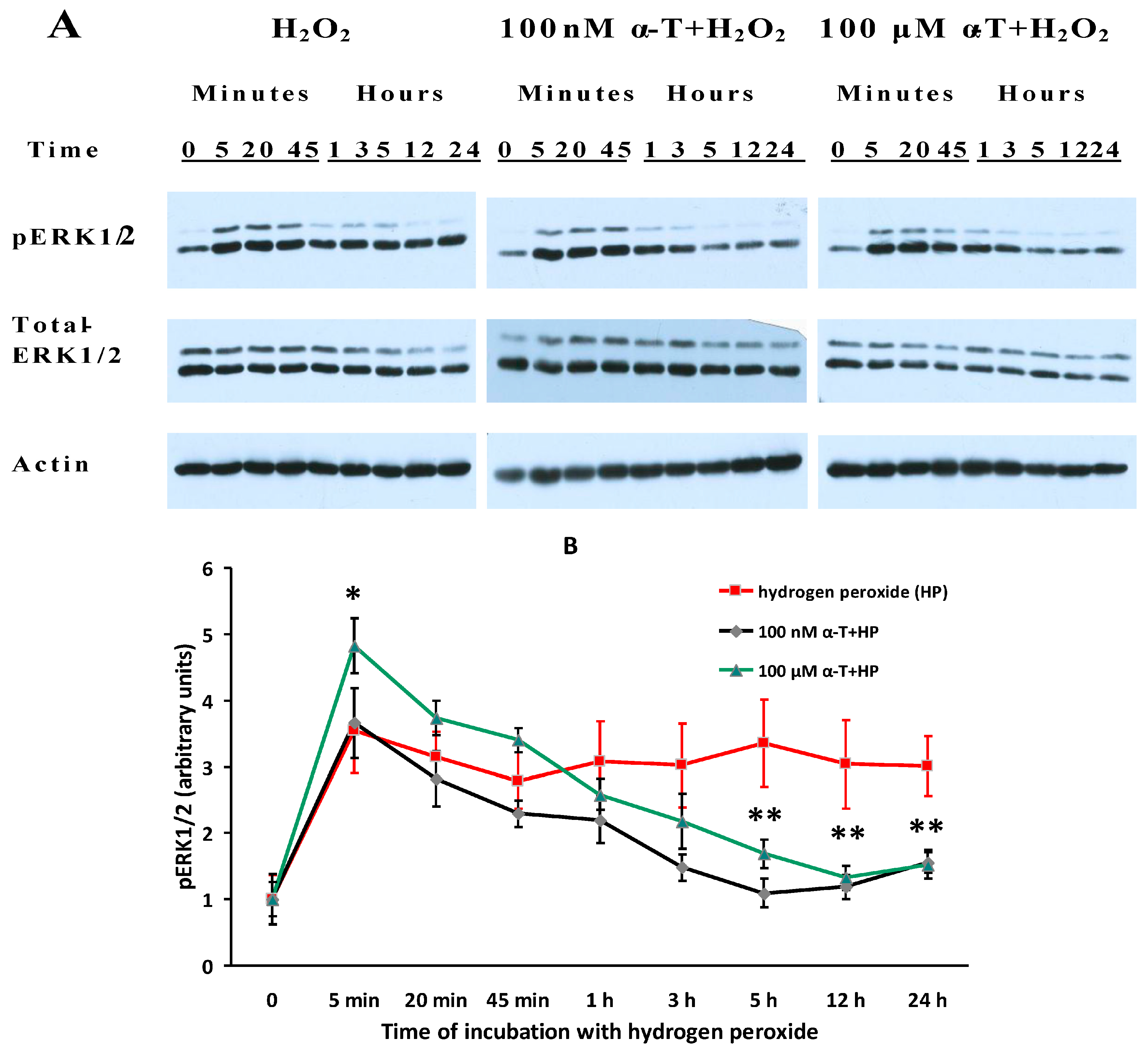
2.7. While α-T Decreases the Time of Long Activation of ERK1/2 in Brain Cortical Neurons Initiated by H2O2, the Effect of 100 nM and 100 μM α-T Is Similar
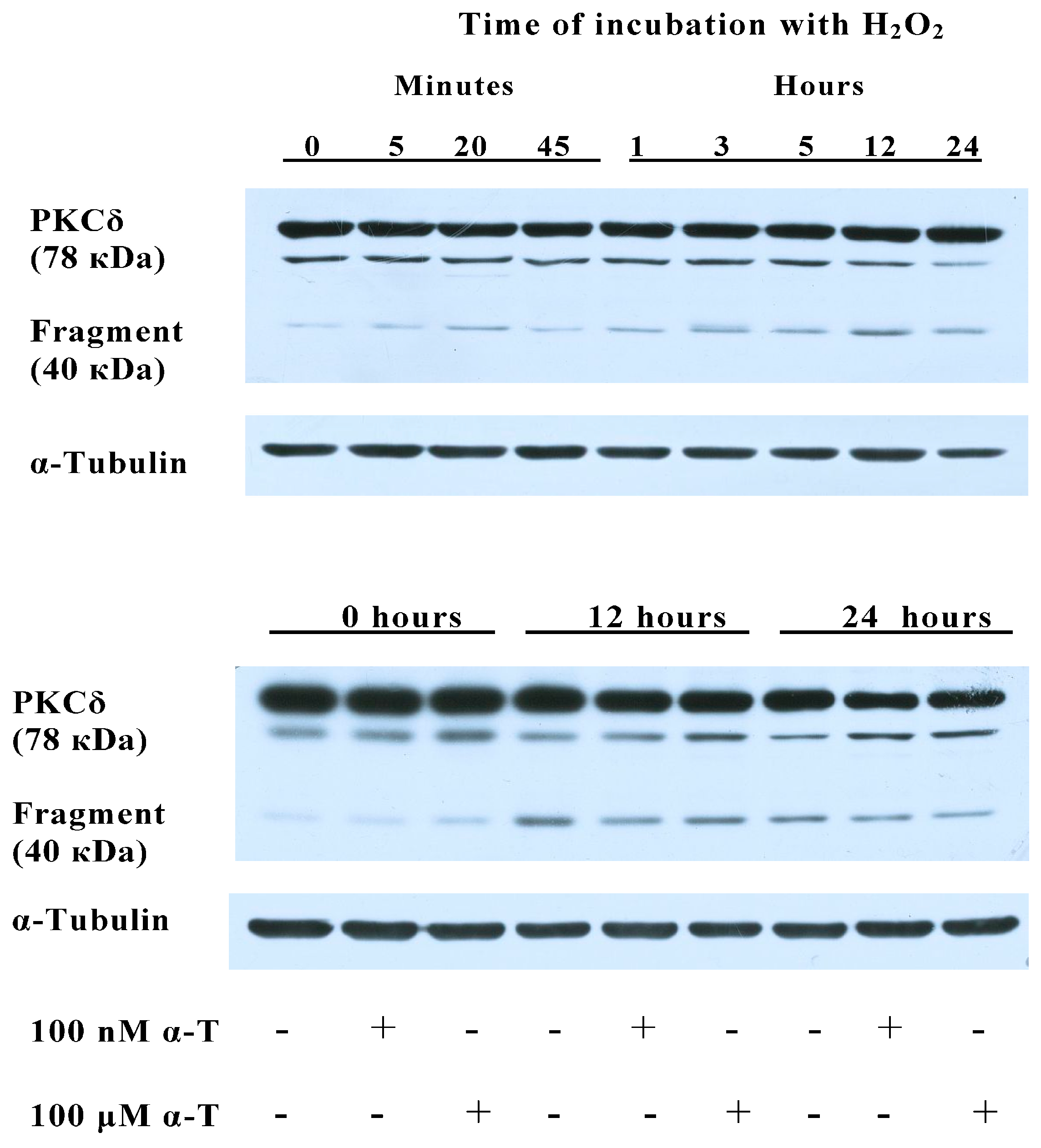
2.8. α-T at 100 μM and 100 nM Concentrations Diminishes the Activation of PKCδ Initiated by H2O2 in Brain Cortical Neurons
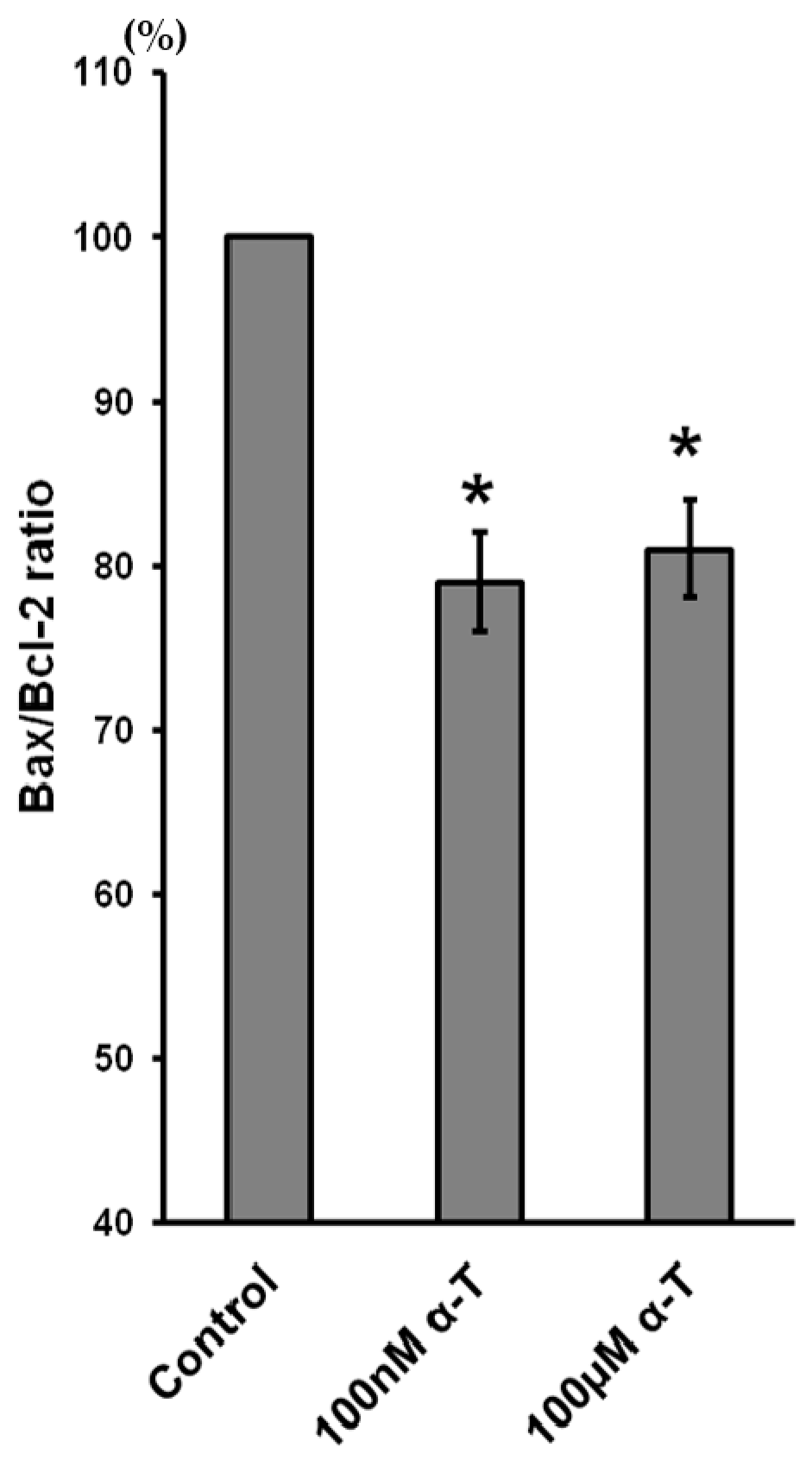
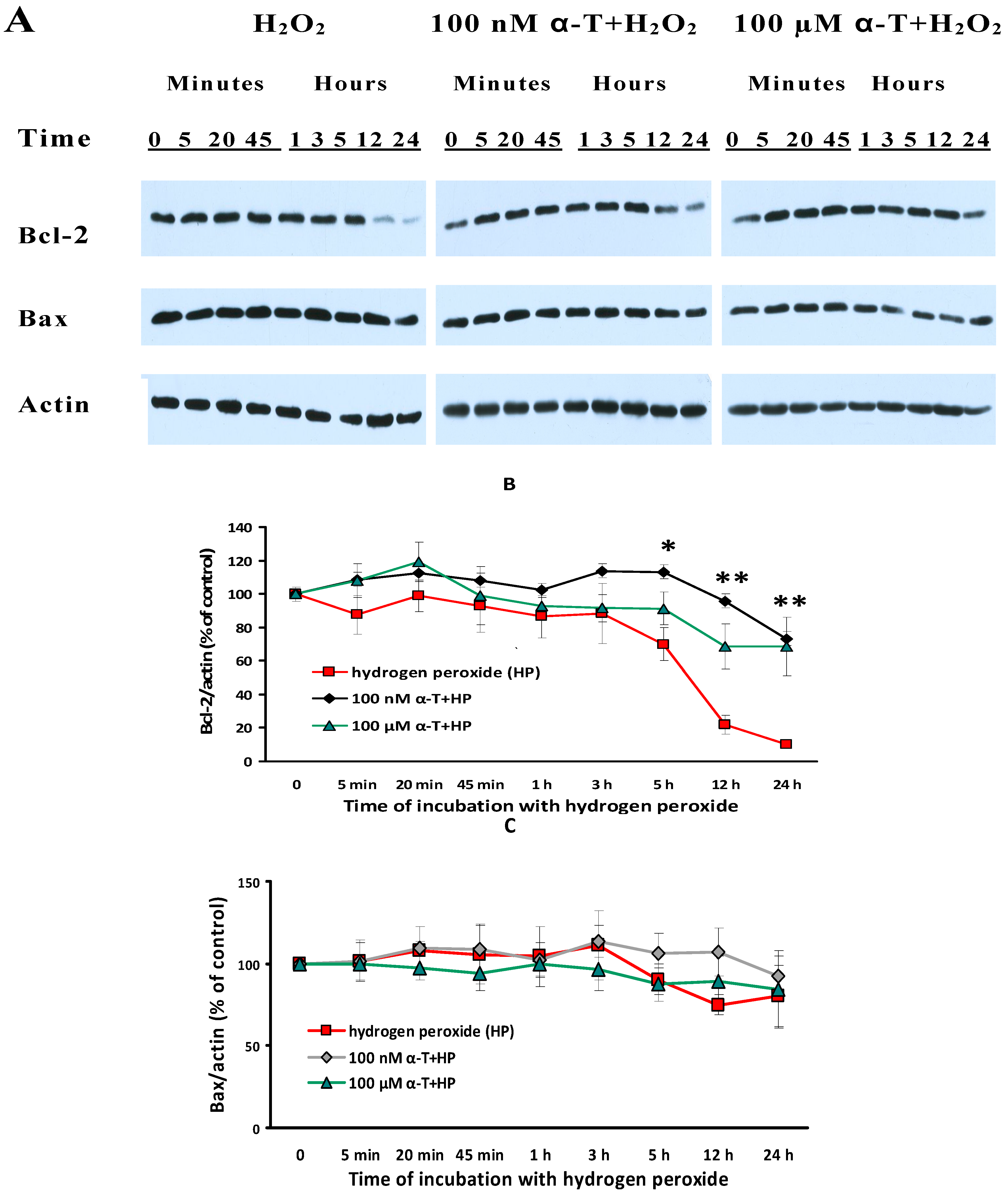
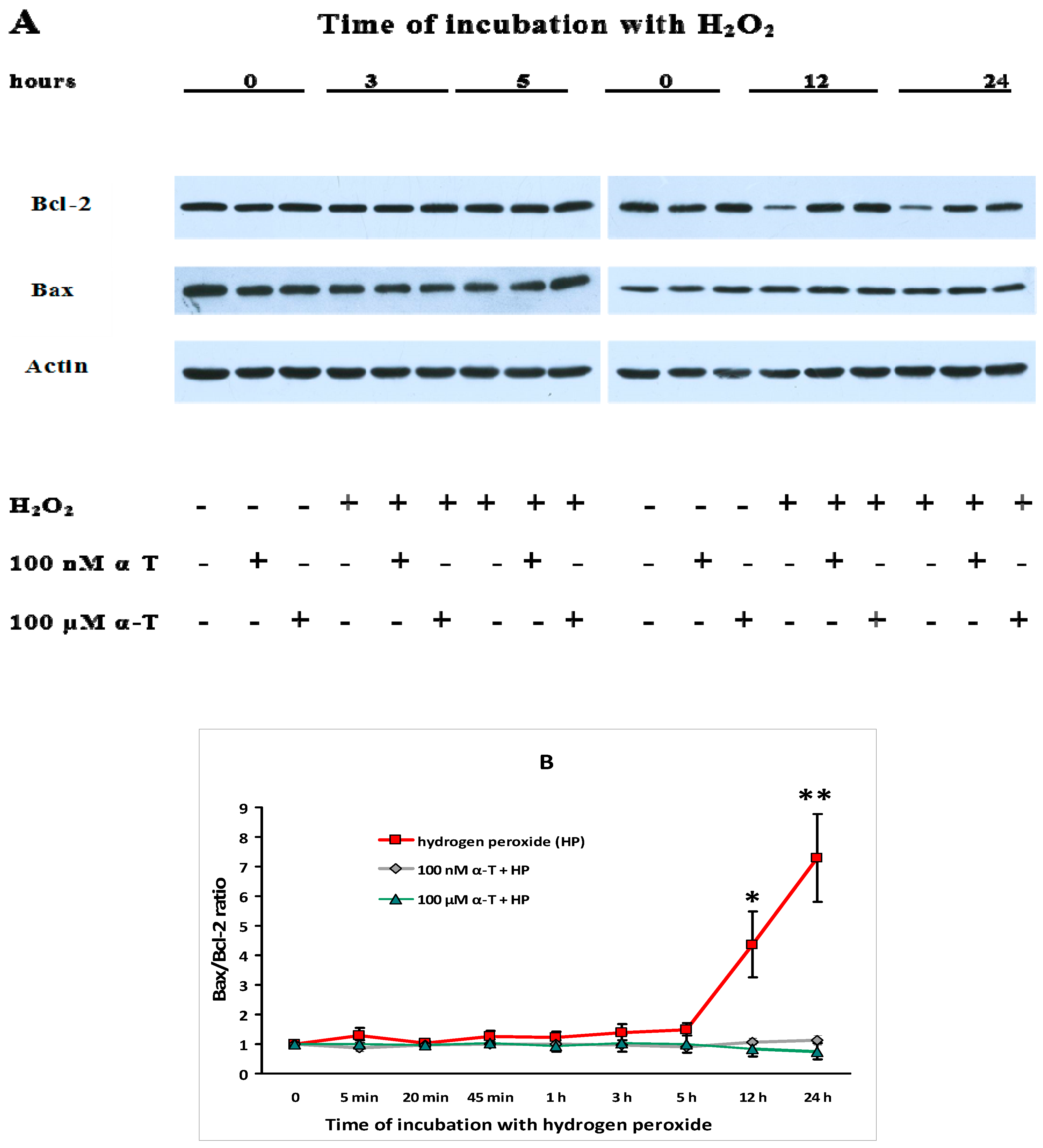
2.9. α-T at Micro- and Nanomolar Concentrations Prevents the Abrupt Decrease of Bcl-2 Level and the Marked Increase of the Bax/Bcl-2 Ratio Initiated by H2O in Brain Cortical Neurons
3. Materials and Methods
3.1. Materials
3.2. Brain Cortical Neurons in Culture
3.3. Assessment of Cell Viability Using the Lactate Dehydrogenase (LDH) Method
3.4. Determination of ROS Accumulation
3.5. Immunoblotting
3.6. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| α-T | α-tocopherol |
| ERK1/2 | extracellular signal-regulated protein kinase |
| PI 3-kinase | phosphatidylinositol 3-kinase |
| Akt | protein kinase B |
| PKC | protein kinase C |
| ROS | reactive oxygen species |
References
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention. Systematic review and meta-analysis. J. Am. Med. Assoc. 2007, 297, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, G.; Nikolova, D.; Gluud, C. Meta-regression analyses, meta-analyses, and trial sequential analyses of the effects of supplementation with beta-carotene, vitamin A, and vitamin E singly or in different combinations on all-cause mortality: Do we have evidence for lack of harm? PLoS ONE 2013, 8, e74558. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Heigel, M.; Weist, J.; Gnyawali, S.; Teplitsky, S.; Roy, S.; Sen, C.K.; Rink, C. Excessive α-tocopherol exacerbates microglial activation and brain injury caused by acute ischemic stroke. FASEB J. 2015, 29, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Vatassery, G.T.; Adityanjee; Quach, H.T.; Smith, W.E.; Kuskowski, M.A.; Melnyk, D. α and γ tocopherols in cerebrospinal fluid and serum from older, male, human subjects. J. Am. Coll. Nutr. 2004, 23, 233–238. [Google Scholar] [CrossRef] [PubMed]
- De Bustos, F.; Jimenez-Jimenez, F.J.; Molina, J.A.; Esteban, J.; Guerrero-Sola, A.; Zurdo, M.; Orti-Pareja, M.; Tallon-Barranco, A.; Gomez-Escalonilla, C.; Ramirez-Ramos, C.; et al. Cerebrospinal fluid levels of α-tocopherol in amyotrophic lateral sclerosis. J. Neural Transm. 1998, 105, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Zingg, J.M. Modulation of signal transduction by vitamin E. Mol. Aspects Med. 2007, 28, 481–506. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Radic. Biol. Med. 2007, 43, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Galli, F.; Azzi, A. Present trends in vitamin E research. Biofactors 2010, 36, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, Y.; Numakawa, T.; Matsumoto, T.; Yagasaki, Y.; Kumamaru, E.; Kunugi, H.; Taguchi, T.; Niki, E. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem. 2006, 97, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Roy, S.; Ryu, H.; Bahaddun, P.; Swaan, P.W.; Ratian, R.R.; Sen, C.K. Molecular basis of vitamin E action: Tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem. 2003, 278, 43508–43515. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.; Parinandi, N.L.; Kotha, S.R.; Roy, S.; Rink, C.; Bibus, D.; Sen, C.K. Nanomolar vitamin E α-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection. J. Neurochem. 2010, 112, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Saito, Y.; Nishio, K.; Akazawa, Y.O.; Yamanaka, K.; Miyama, A.; Yoshida, Y.; Noguchi, N.; Niki, E. Cytoprotective effects of vitamin E homologues against glutamate-induced cell death in immature primary cortical neuron cultures: Tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic. Biol. Med. 2010, 49, 1542–1549. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.O.; Sokolova, T.V.; Bayunova, L.V.; Vlasova, Y.A.; Rychkova, M.P.; Avrova, N.F. α-Tocopherol at nanomolar concentration protects PC12 cells from hydrogen peroxide-induced death and modulates protein kinase activities. Int. J. Mol. Sci. 2012, 13, 11543–11568. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Bianchi, S.; Federico, A. Ataxia with vitamin E deficiency: Update of molecular diagnosis. Neurol. Sci. 2010, 31, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Ames, D.; Ritchie, C. Antioxidants and Alzheimer’s disease: Time to stop feeding vitamin E to dementia patients? Int. Psychogeriatr. 2007, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cuddihy, S.L.; Ali, S.S.; Musiec, E.S.; Lucero, J.; Kopp, S.J.; Morrow, J.D.; Dugan, L.L. Prolonged α-tocopherol deficiency decreases oxidative stress and unmasks α-tocopherol-dependent regulation of mitochondrial function in the brain. J. Biol. Chem. 2008, 283, 6915–6924. [Google Scholar] [CrossRef] [PubMed]
- De Jesus Ferreira, M.C.; Crouzin, N.; Barbanel, G.; Cohen-Solal, C.; Recasens, M.; Vignes, M.; Guiramand, J. A transient treatment of hippocampal neurons with α-tocopherol induces a long-lasting protection against oxidative damage via a genomic action. Free Radic. Biol. Med. 2005, 39, 1009–1020. [Google Scholar] [CrossRef] [PubMed]
- Crouzin, N.; Ferreira, M.C.; Cohen-Solal, C.; Barbanel, G.; Guiramand, J.; Vignes, M. Neuroprotection induced by vitamin E against oxidative stress in hippocampal neurons: Involvement of TRPV1 channels. Mol. Nutr. Food Res. 2010, 54, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Obitsu, S.; Ohta, S.; Matsunami, K.; Otsuka, H.; Teshima, R. Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by α-tocopherol and MEK inhibition. J. Biol. Chem. 2010, 285, 13079–13091. [Google Scholar] [CrossRef] [PubMed]
- Susarla, B.T.S.; Robinson, M.B. Rottlerin, an inhibitor of protein kinase Cδ (PKCδ), inhibits astrocytic glutamate transport activity and reduces GLAST immunoreactivity by a mechanism that appears to be PKCδ-independent. J. Neurochem. 2003, 86, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.N.; Wang, Y.J.; Lin, M.W. Potent stimulation of large-conductance Ca2+-actiated K+ channels by rottlerin, an inhibitor of protein kinase C-δ, in pituitary tumor (GH3) cells and in cortical neuronal (HCN-1A) cells. J. Cell Physiol. 2007, 210, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, Y.A.; Avrova, N.F. Antioxidant effects of α-tocopherol at nanomolar concentrations: The role of the modulation of the activity of signaling systems. Neurochem. J. 2010, 4, 178–184. [Google Scholar] [CrossRef]
- Ziegler, C.G.; Sicard, F.; Sperber, S.; Ehrhart-Bornstein, M.; Bornstein, S.R.; Krug, A.W. DHEA reduces NGF-mediated cell survival in serum-deprived PC12 cells. Ann. N. Y. Acad. Sci. 2006, 1073, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Williams, R.J.; Spencer, J.P. Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J. Neurochem. 2007, 103, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ren, M.; Jiang, H.; Cui, S.; Wang, S.; Jiang, H.; Qi, Y.; Wang, J.; Wang, X.; Dong, G.; Leeds, P.; Chuang, D.M.; Feng, H. Downregulated AEG-1 together with inhibited PI3K/Akt pathway is associated with reduced viability of motor neurons in an ALS model. Mol. Cell Neurosci. 2015, 68, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.O.; Sokolova, T.V.; Vlasova, Y.A.; Furaev, V.V.; Rychkova, M.P.; Avrova, N.F. GM1 ganglioside activates ERK1/2 and Akt downstream of Trk tyrosine kinase and protects PC12 cells against hydrogen peroxide toxicity. Neurochem. Res. 2014, 39, 2262–2275. [Google Scholar] [CrossRef] [PubMed]
- Murata, H.; Ihara, Y.; Nakamura, H.; Yodoi, J.; Sumikawa, K.; Kondo, T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J. Biol. Chem. 2003, 278, 50226–50233. [Google Scholar] [CrossRef] [PubMed]
- Daou, G.B.; Srivastava, A.K. Reactive oxygen species mediate Endothelin-1-induced activation of ERK1/2, PKB, and Pyk2 signaling, as well as protein synthesis, in vascular smooth muscle cells. Free Radic. Biol. Med. 2004, 37, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ichikawa, S.; Tani, C.; Zhu, B.; Tada, M.; Shimoishi, Y.; Murata, Y.; Nakamura, Y. Docosahexaenoic acid induces ERK1/2 activation and neuritogenesis via intracellular reactive oxygen species production in human neuroblastoma SH-SY5Y cells. Biochim. Biophys. Acta 2009, 1791, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Kulebyakin, K.; Karpova, L.; Lakonsteva, E.; Krasavin, M.; Boldyrev, A. Carnosine protects neurons against oxidative stress and modulates the time profile of MAPK cascade signaling. Amino Acids 2012, 43, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Azzi, A.; Breyer, I.; Feher, M.; Ricciarelli, R.; Stocker, A.; Zimmer, S.; Zingg, J. Nonantioxidant functions of α-tocopherol in smooth muscle cells. J. Nutr. 2001, 131, 378S–381S. [Google Scholar] [PubMed]
- Yano, T.; Yajima, S.; Hagiwara, K.; Kumadaki, I.; Yano, Y.; Otani, S.; Uchida, M.; Ichikawa, T. Vitamin E inhibits cell proliferation and the activation of extracellular signal-regulated kinase during the promotion phase of lung tumorigenesis irrespective of antioxidative effect. Carcinogenesis 2000, 21, 2129–2133. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Aizenman, E. ERK signaling leads to mitochondrial dysfunction in extracellular zinc-induced neurotoxicity. J. Neurochem. 2010, 114, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.; Samarasinghe, R.; Knoch, M.E.; Lewis, M.E.; Aizenman, E.; DeFranco, D.B. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellar signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol. Pharmacol. 2008, 74, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; DeFranco, D.B. Opposing roles for ERK 1/2 in neuronal oxidative toxicity: Distinct mechanisms of ERK 1/2 action at early versus late phases of oxidative stress. J. Biol. Chem. 2006, 281, 16436–16442. [Google Scholar] [CrossRef] [PubMed]
- Namura, S.; Iihara, K.; Takami, S.; Nagata, I.; Kikuchi, H.; Matsushita, K.; Moskowitz, M.A.; Bonventre, J.V.; Alessandrini, A. Intravenous administration of MEK inhibitor UO126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2001, 98, 11569–11574. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Xu, L.; Rozanski, D.J.; Sugawara, T.; Chan, P.H.; Trzaskos, J.M.; Feuerstein, G.Z. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: Exploration of potential mechanism associated with apoptosis. J. Pharmacol. Exp. Therap. 2003, 304, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Liang, C.L.; Liliang, P.C.; Yang, S.-H.; Cho, C.L.; Weng, H.C.; Tsai, Y.-D.; Wang, K.W.; Chen, H.J. Inhibition of extracellular signal-regulated kinases 1/2 provides neuroprotection in spinal cord ischemia/reperfusion injury in rats: Relationship with the nuclear factor-kB-regulated anti-apoptotic mechanisms. J. Neurochem. 2010, 114, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Hanrott, K.; Gudmunsen, L.; O’Neill, M.J.; Wonnacott, S. 6-Hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of proteinkinase Cδ. J. Biol. Chem. 2006, 281, 5373–5382. [Google Scholar] [CrossRef] [PubMed]
- Carvour, M.; Song, C.; Kaul, S.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A. Chronic low-dose oxidative stress induces caspase-3-dependent PKCδ proteolyticactivation and apoptosis in a cell culture model of dopaminergic neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1139, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Yamanouchi, D.; Esbona, K.; Kamiya, K.; Zhang, F.; Kent, K.C.; Liu, B. Caspase-mediated protein kinase C-δ cleavage is necessary for apoptosis of vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Ferri, P.; Cecchini, T.; Ambrogini, P.; Betti, M.; Cuppini, R.; del Grande, P.; Ciaroni, S. α-Tocopherol affects neuronal plasticity in adult rat dentate gyrus: The possible role of PKCδ. J. Neurobiol. 2006, 66, 793–810. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Neuroprotective effect of protein kinase Cδ inhibitor rottlerin in cell culture and animal models of Parkinson's disease. J. Pharmacol. Exp. Ther. 2007, 322, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Latchoumycandane, C.; Anantharam, V.; Jin, H.; Kanthasamy, A.; Kanthasamy, A. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson’s disease. Toxicol. Appl. Pharmacol. 2011, 256, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Kanthasamy, A.; Harischandra, D.S.; Kondru, N.; Ghosh, A.; Panicker, N.; Anantharam, V.; Rana, A.; Kanthasamy, A.G. Histone hyperacetylation up-regulates protein kinase Cδ in dopaminergic neurons to induce cell death: Relevance to epigenetic mechanisms of neurodegeneration in Parkinson disease. J. Biol. Chem. 2014, 289, 34743–34767. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Kanthasamy, A.; Song, C.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. Proteasome inhibitor-induced apoptosis is mediated by positive feedback amplification of PKCδ proteolytic activation and mitochondrial translocation. J. Cell. Mol. Med. 2008, 12, 2467–2481. [Google Scholar] [CrossRef] [PubMed]
- Basu, A. Involvement of protein kinase C-δ in DNA-damage induced apoptosis. J. Cell. Mol. Med. 2003, 7, 341–350. [Google Scholar] [CrossRef] [PubMed]
- DeVries-Seimon, T.A.; Ohm, A.M.; Humphries, M.J.; Reyland, M.T. Induction of apoptosis is driven by nuclear retention of protein kinas Cδ. J. Biol. Chem. 2007, 282, 22307–22314. [Google Scholar] [CrossRef] [PubMed]
- Sitailo, L.A.; Tibudan, S.S.; Denning, M.F. Bax activation and induction of apoptosis in human keratinocytes by the protein kinase C-δ catalytic domain. Investig. Dermatol. 2004, 123, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Betti, M.; Ambrogini, P.; Minelli, A.; Floridi, A.; Lattanzi, D.; Ciuffoli, S.; Bucherelli, C.; Prospero, E.; Frontini, A.; Santarelli, L.; et al. Maternal dietary load of α-tocopherol depress protein kinase C signaling and synaptic plasticity in rat postnatal developing hippocampus and promotes permanent deficits in adult offspring. J. Nutr. Biochem. 2011, 22, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Salucci, S.; Ambrogini, P.; Lattanzi, D.; Betti, M.; Gobbi, P.; Galati, C.; Galli, F.; Cuppini, R.; Minelli, A. Maternal dietary loads of α-tocopherol increase synapse density and glial synaptic coverage in the hippocampus of adult offspring. Eur. J. Histochem. 2014, 58, 2355. [Google Scholar] [CrossRef] [PubMed]
- Then, S.M.; Mazlan, M.; Top, G.M.; Ngah, W.Z.W. Is vitamin E toxic to neuron cells? Cell Mol. Neurobiol. 2009, 29, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria inapoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.W.; Chen, J.C.; Lai, T.Y.; Yang, J.S.; Weng, S.W.; Ma, Y.S.; Lin, H.Y.; Wu, R.S.; Wu, K.C.; Wood, W.G.; et al. Gypenosides suppress growth of human oral cancer SAS cells in vitro and in a murine xenograft model: The role of apoptosis mediated by caspase-dependent and caspase-independent pathways. Integr. Cancer Ther. 2012, 11, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Chang, A.Y.; Wang, L.L.; Ou, C.C.; Chan, S.H. Protein kinase C-dependent mitochondrial translocation of proapoptotic protein Bax on activation of inducible nitric oxide synthase in rostral ventrolateral medulla mediates cardiovascular depression during experimental endotoxemia. Mol. Pharmacol. 2007, 71, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, B.; Galluzzi, L.; Kepp, O.; Kroemer, G. Adenine nucleotide translocase: A component of the phylogenetically conserved cell death machinery. Cell Death Differ. 2009, 16, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B.; et al. Bax regulates primary necrosisthrough mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; Fiscum, G. Redox mechanisms of cytoprotection by Bcl-2. Antioxid. Redox Signal. 2005, 7, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Hsuan, S.L.; Klintworth, H.M.; Xia, Z. Basic fibroblast growth factor protects against rotenone-induced dopaminergic cell death through activation of extracellular signal-regulated kinases 1/2 and phosphatidylinositol-3 kinase pathways. J. Neurosci. 2006, 26, 4481–4491. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, O. Novel neuroprotective mechanism of action of rasagiline is associated with its propargyl moiety: Interaction of Bcl-2 family members with PKC pathway. Ann. N. Y. Acad. Sci. 2005, 1053, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Dichter, M.A. Rat cortical neurons in cell culture: Culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res. 1978, 149, 279–293. [Google Scholar] [CrossRef]
- Mironova, E.V.; Evstratova, A.A.; Antonov, S.M. A fluorescence vital assay for the recognition and quantification of excitotoxic cell death by necrosis and apoptosis using confocal microscopy on neurons in culture. J. Neurosci. Methods 2007, 163, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Kussmaul, L.; Hamprecht, B. Detoxification of exogenous hydrogen peroxide and organic hydroperoxides by cultured astroglial cells assessed by microtiter plate assay. Brain Res. Protoc. 1998, 2, 223–228. [Google Scholar] [CrossRef]
- Nagasaki, H.; Nakashima, A.; Kaneko, Y.S.; Kodani, Y.; Takayanagi, T.; Itoh, M.; Kondo, K.; Nagatsu, T.; Hamada, Y.; Ota, M.; et al. Aripiprazole increases NADPH level in PC12 cells: The role of NADPH oxidase. J. Neural Transm. 2014, 121, 91–103. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| α-T Concentration | 100 μM α-T | 10 μM α-T | 1 μM α-T | 100 nM α-T | 10 nM α-T | 1 nM α-T |
|---|---|---|---|---|---|---|
| Rescue rates (%) | 64.3 ± 7.2 * | 67.4 ± 11.9 * | 60.0 ± 11.4 * | 52.5 ± 7.4 *x | 27.3 ± 5.1 *# | 5.1 ± 2.9 |
| Sample | Rescue Rates of α-T, % | Sample | Rescue Rates of α-T, % |
|---|---|---|---|
| 100 nM α-T | 52.4 ± 13.1 ** | 100 μM α-T | 63.35 ± 12.6 ** |
| 100 nM α-T + 10 µM SL327 | 31.35 ± 11.1 *,# | 100 μM α-T + 10 μM SL327 | 36.9 ± 12.3 * |
| 100 nM α-T + 50 µM LY294002 | 10.1 ± 4.6 x | 100 μM α-T + 50 μM LY294002 | 27.6 ± 14.7 # |
| 100 nM α-T | 50.0 ± 7.5 ** | 100 μM α-T | 52.2 ± 6.4 ** |
| 100 nM α-T + 5 µM rottlerin | 20.9 ± 4.6 **,x | 100 μM α-T + 5 μM rottlerin | 33.6 ± 4.8 **,x |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharova, I.O.; Sokolova, T.V.; Vlasova, Y.A.; Bayunova, L.V.; Rychkova, M.P.; Avrova, N.F. α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress. Int. J. Mol. Sci. 2017, 18, 216. https://doi.org/10.3390/ijms18010216
Zakharova IO, Sokolova TV, Vlasova YA, Bayunova LV, Rychkova MP, Avrova NF. α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress. International Journal of Molecular Sciences. 2017; 18(1):216. https://doi.org/10.3390/ijms18010216
Chicago/Turabian StyleZakharova, Irina O., Tatiana V. Sokolova, Yulia A. Vlasova, Liubov V. Bayunova, Maria P. Rychkova, and Natalia F. Avrova. 2017. "α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress" International Journal of Molecular Sciences 18, no. 1: 216. https://doi.org/10.3390/ijms18010216
APA StyleZakharova, I. O., Sokolova, T. V., Vlasova, Y. A., Bayunova, L. V., Rychkova, M. P., & Avrova, N. F. (2017). α-Tocopherol at Nanomolar Concentration Protects Cortical Neurons against Oxidative Stress. International Journal of Molecular Sciences, 18(1), 216. https://doi.org/10.3390/ijms18010216