
Decreased Sp1 Expression Mediates Downregulation of SHIP2 in Gastric Cancer Cells

Abstract
:1. Introduction
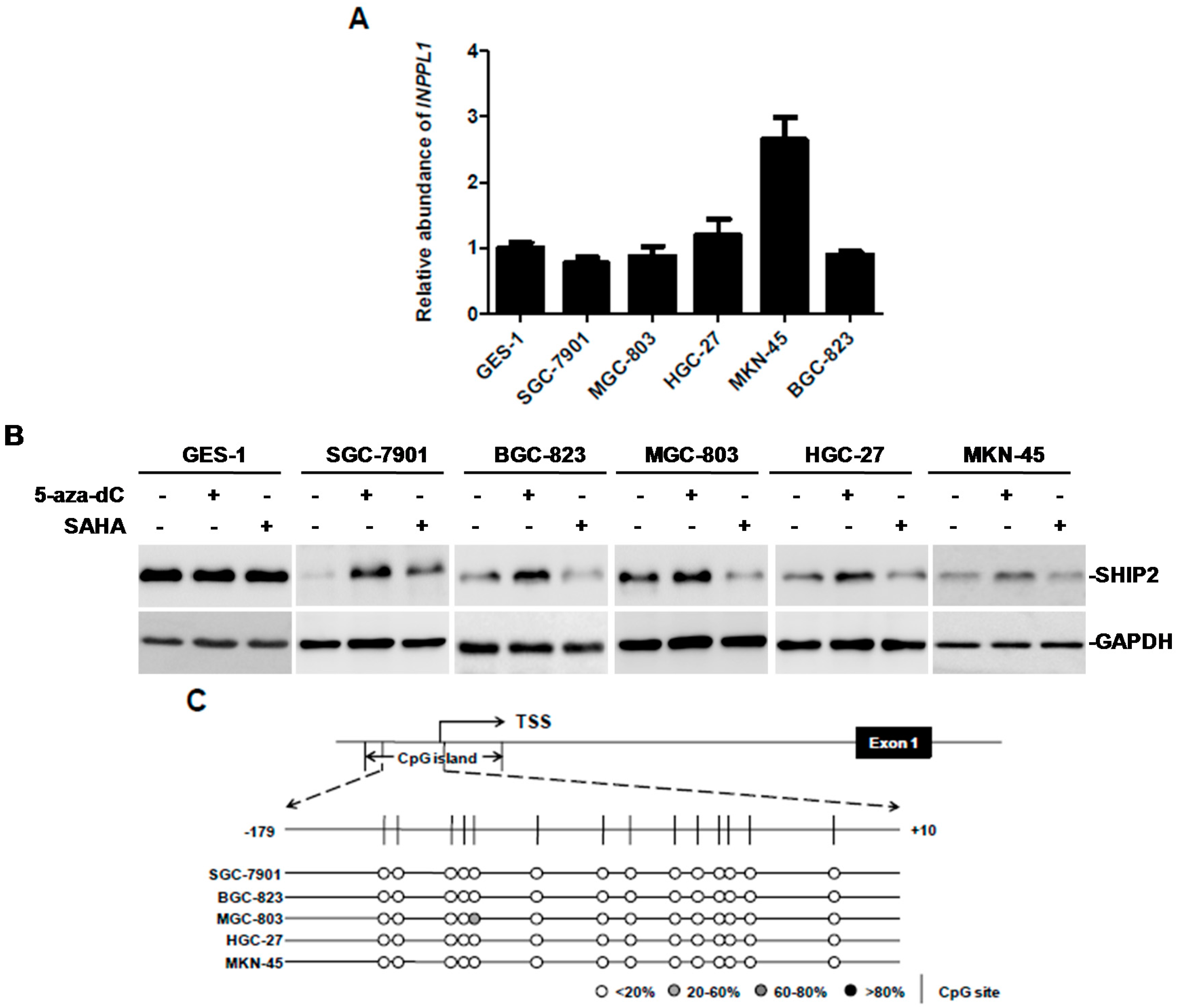
2. Results
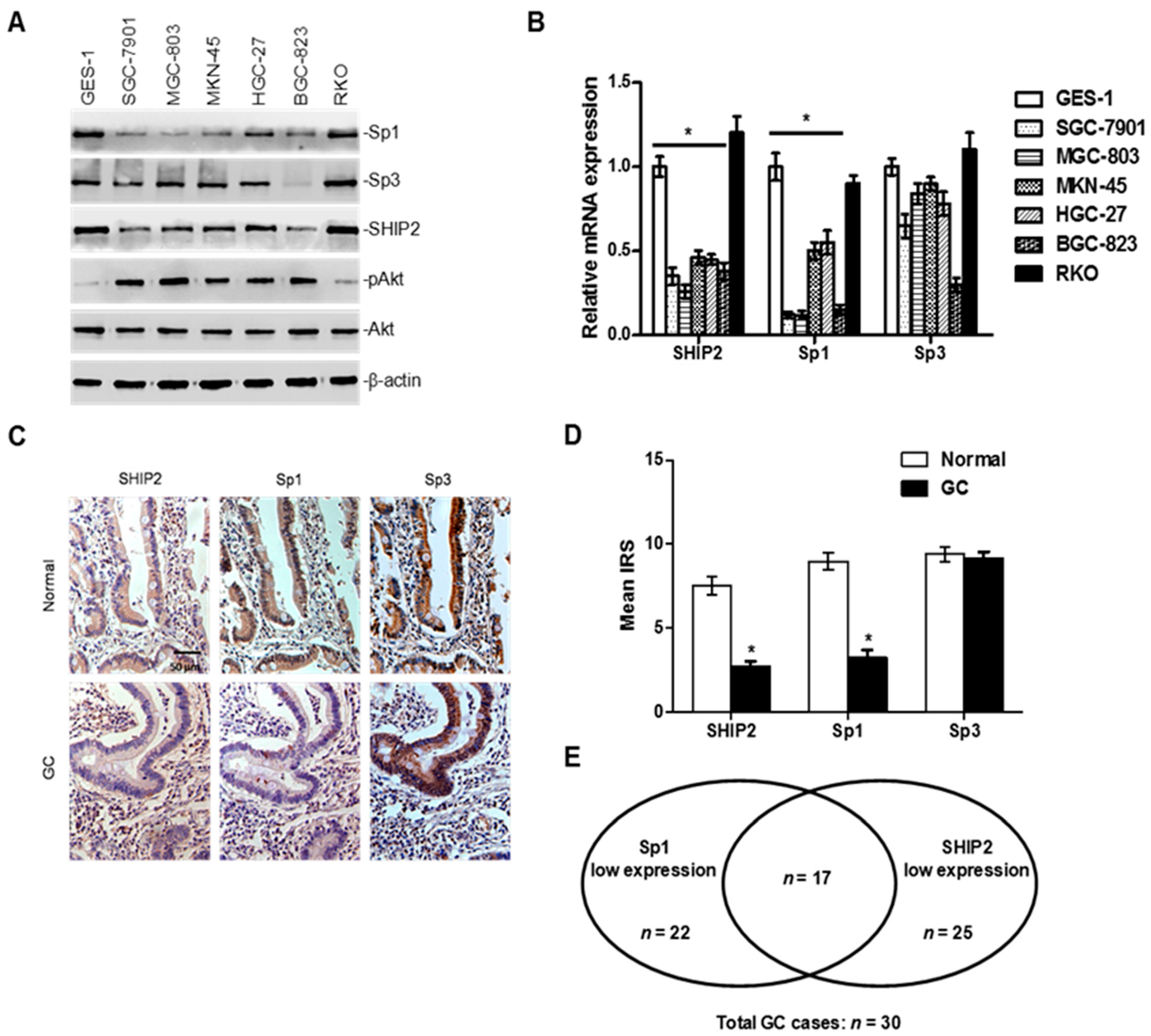
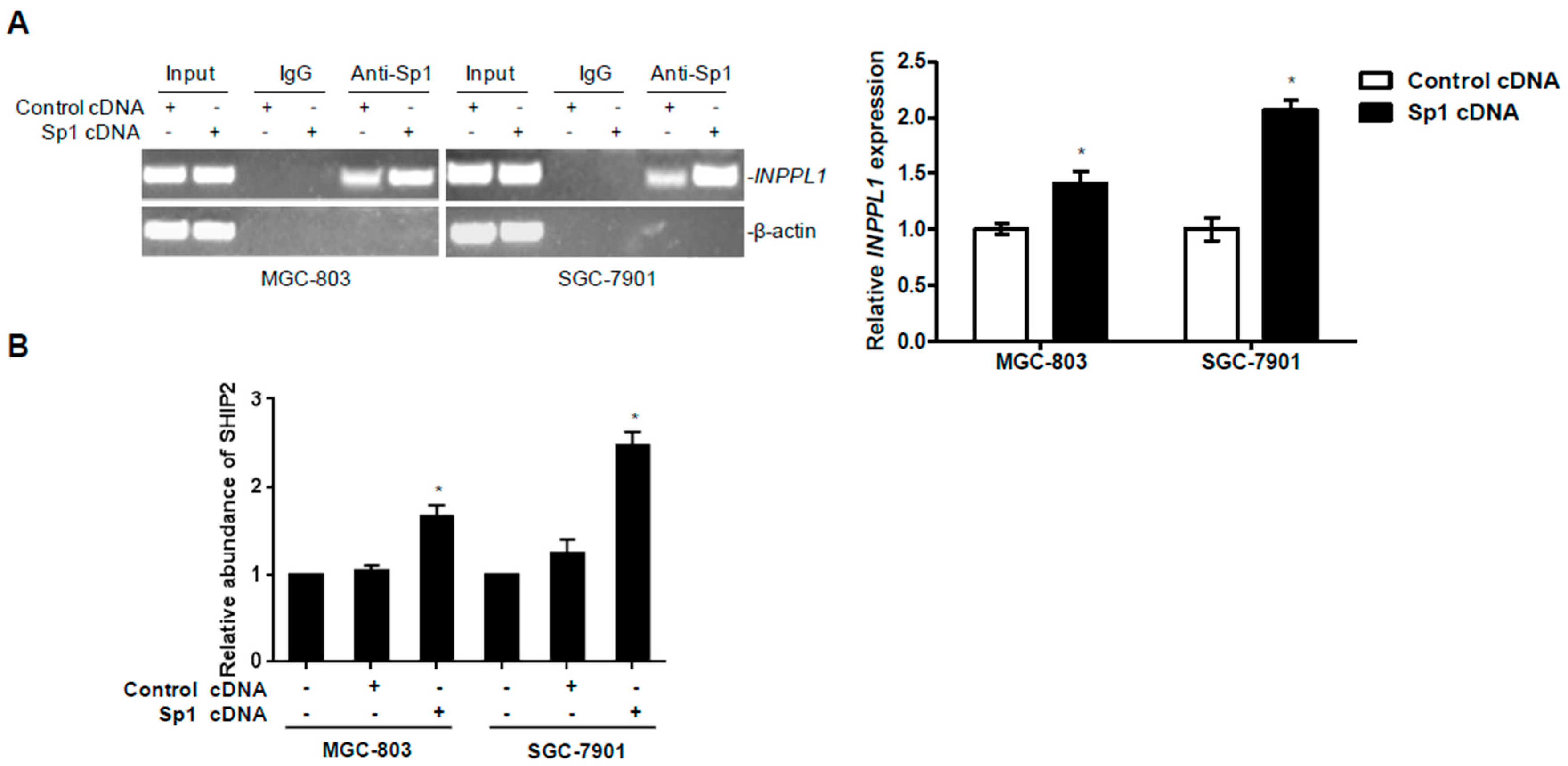
2.1. SHIP2 Is Regulated by Transcription Factor Sp1
2.2. Decreased Sp1 Expression Parallels the Downregulation of SHIP2 in GC
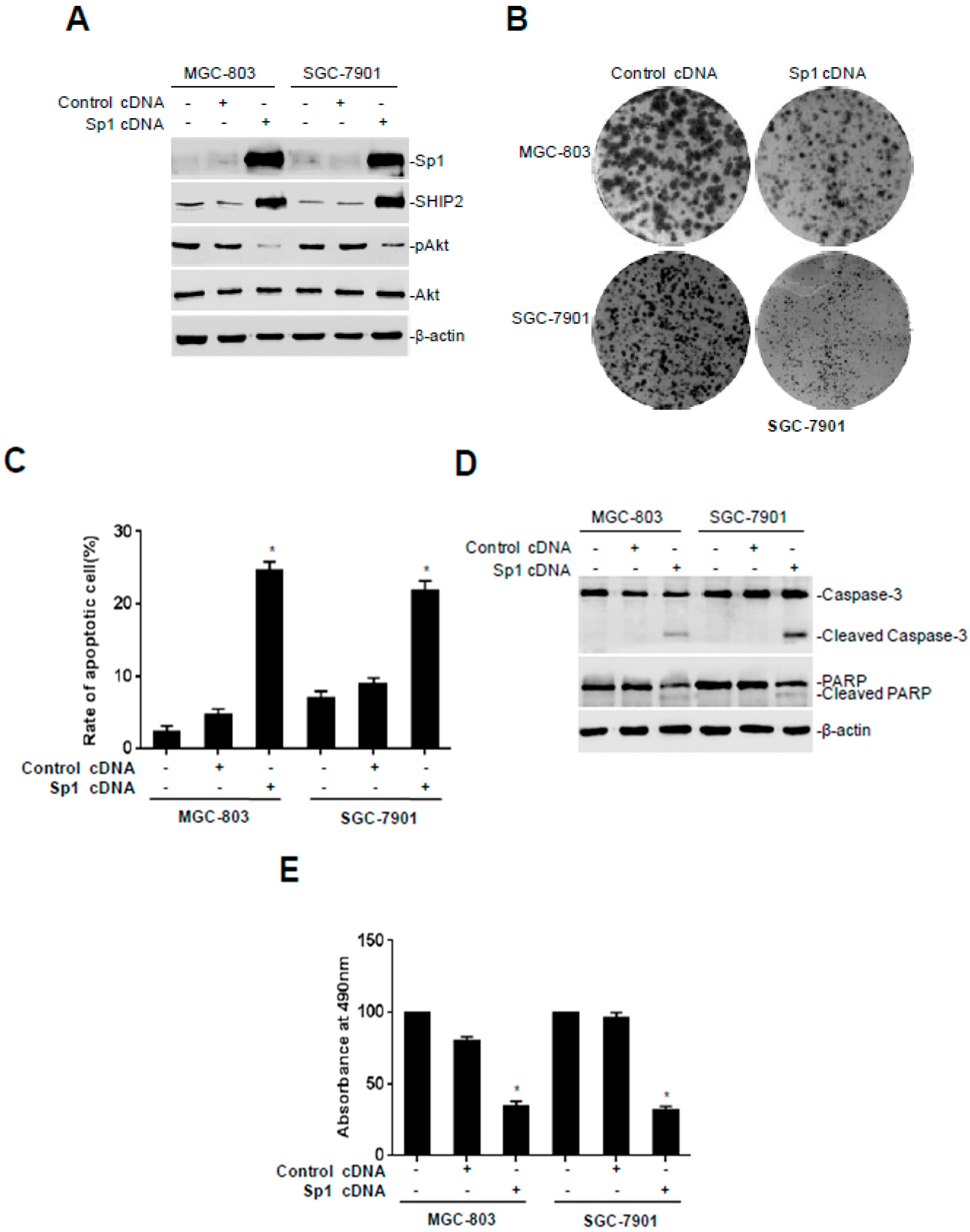
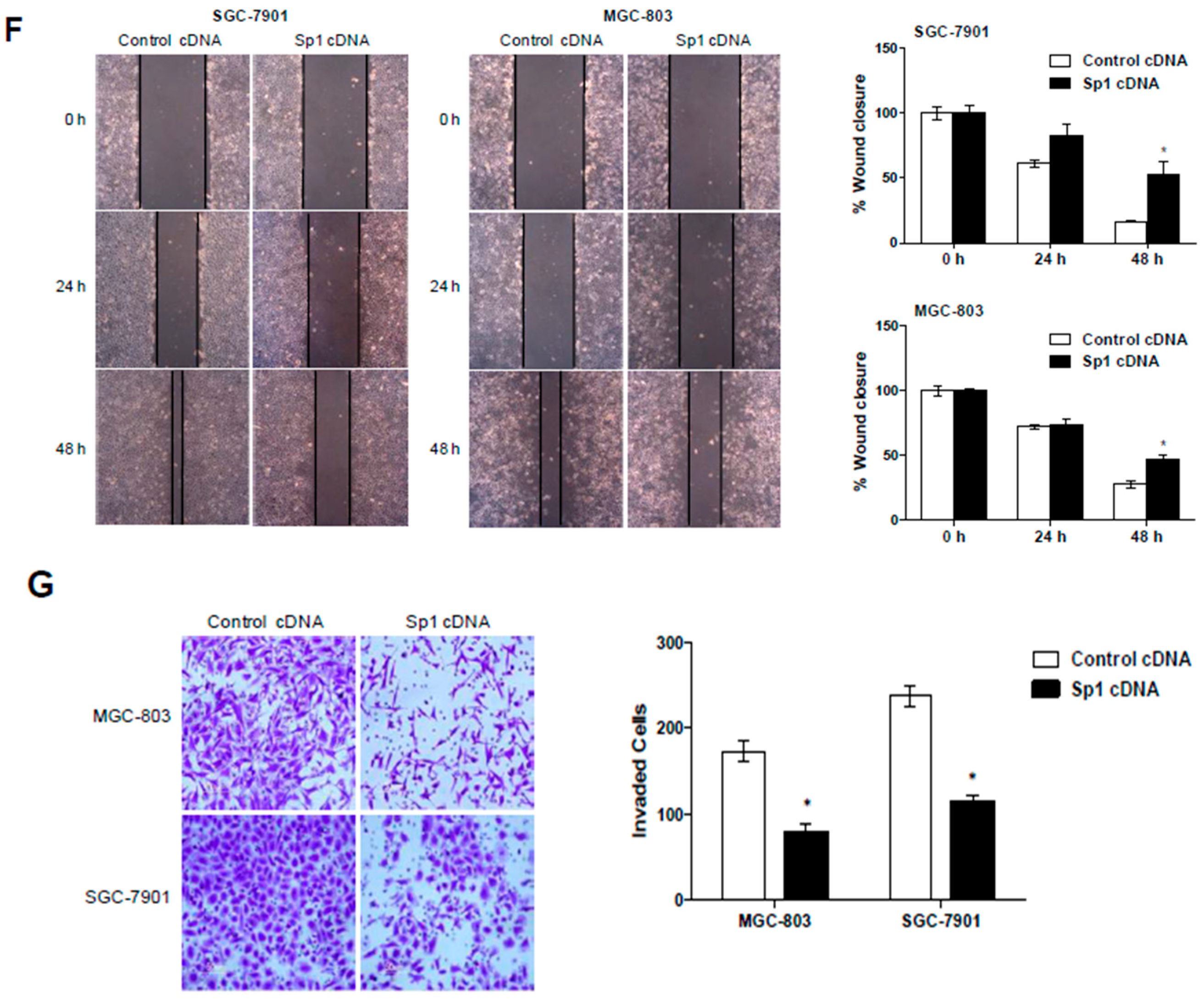
2.3. Overexpression of Sp1 Inhibits Malignant Behaviors of GC Cells
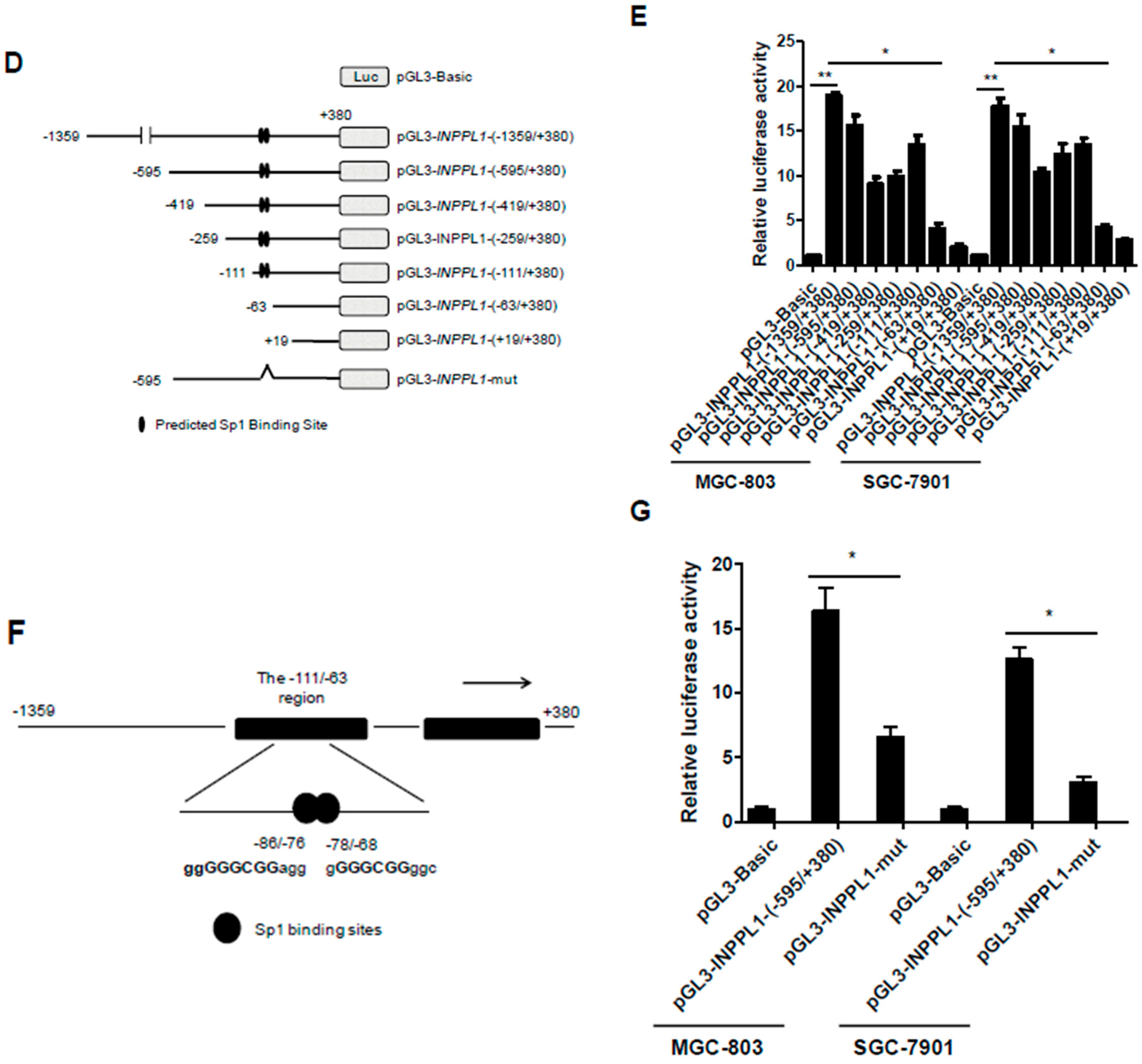
2.4. Overexpression of Sp1 Enhances the Transcriptional Activity of SHIP2 Promoter
3. Discussion
4. Materials and Methods
4.1. Human Tissues, Cell Lines, and Cell Culture
4.2. Immunohistochemistry (IHC)
4.3. Western Blot
4.4. Plasmid Vectors and Transfection
4.5. Cell Proliferation and Colony Formation Assays
4.6. Apoptosis Assay
4.7. Wound Healing and Matrigel Invasion Assays
4.8. RNA Isolation and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR)
4.9. Chromatin Immunoprecipitation Assays
4.10. Dual Luciferase Reporter Assays
4.11. Exon Sequencing
4.12. Bisulfite Genomic Sequencing (BGS) Analysis
4.13. qPCR Analysis of Copy Number Variations
4.14. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.; Yeoh, K.G. Genetics and molecular pathogenesis of gastric adenocarcinoma. Gastroenterology 2015, 149, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Wang, L.; Ajani, J.; Xie, K. Molecular basis of gastric cancer development and progression. Gastric Cancer 2004, 7, 61–77. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Ge, Y.M.; Xiao, M.M.; Guo, L.M.; Li, Q.; Hao, J.Q.; Da, J.; Hu, W.L.; Zhang, X.D.; Xu, J.; et al. Suppression of SHIP2 contributes to tumorigenesis and proliferation of gastric cancer cells via activation of Akt. J. Gastroenterol. 2016, 51, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Elong Edimo, W.; Schurmans, S.; Roger, P.P.; Erneux, C. SHIP2 signaling in normal and pathological situations: Its impact on cell proliferation. Adv. Biol. Regul. 2014, 54, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Pesesse, X.; Deleu, S.; de Smedt, F.; Drayer, L.; Erneux, C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase ship. Biochem. Biophys. Res. Commun. 1997, 239, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Prasad, N.K.; Tandon, M.; Badve, S.; Snyder, P.W.; Nakshatri, H. Phosphoinositol phosphatase SHIP2 promotes cancer development and metastasis coupled with alterations in EGF receptor turnover. Carcinogenesis 2008, 29, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Rajadurai, C.V.; Havrylov, S.; Coelho, P.P.; Ratcliffe, C.D.; Zaoui, K.; Huang, B.H.; Monast, A.; Chughtai, N.; Sangwan, V.; Gertler, F.B.; et al. 5′-inositol phosphatase SHIP2 recruits MENA to stabilize invadopodia for cancer cell invasion. J. Cell Biol. 2016, 214, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Fan, W.; Pu, X.; Ni, H.; Zhang, W.; Chang, F.; Gong, L.; Xiong, L.; Wang, J.; Gu, X. Elevated expression of SHIP2 correlates with poor prognosis in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2013, 6, 2185–2191. [Google Scholar] [PubMed]
- Fu, M.; Gu, X.; Ni, H.; Zhang, W.; Chang, F.; Gong, L.; Chen, X.; Li, J.; Qiu, L.; Shi, C.; et al. High expression of inositol polyphosphate phosphatase-like 1 associates with unfavorable survival in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2013, 6, 2515–2522. [Google Scholar] [PubMed]
- Prasad, N.K.; Tandon, M.; Handa, A.; Moore, G.E.; Babbs, C.F.; Snyder, P.W.; Bose, S. High expression of obesity-linked phosphatase SHIP2 in invasive breast cancer correlates with reduced disease-free survival. Tumour. Biol. 2008, 29, 330–341. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Fu, M.; Ding, Y.; Weng, Y.; Fan, W.; Pu, X.; Ge, Z.; Zhan, F.; Ni, H.; Zhang, W.; et al. High SHIP2 expression indicates poor survival in colorectal cancer. Dis. Markers 2014, 2014, 218968. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Ryan, D.G.; Getsios, S.; Oliveira-Fernandes, M.; Fatima, A.; Lavker, R.M. Microrna-184 antagonizes microrna-205 to maintain SHIP2 levels in epithelia. Proc. Natl. Acad. Sci. USA 2008, 105, 19300–19305. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.; Wong, M.; Brandts, C.; Reilly, L.; Dean, N.M.; Cowsert, L.M.; Moodie, S.; Stokoe, D. 5′-phospholipid phosphatase SHIP-2 causes protein kinase b inactivation and cell cycle arrest in glioblastoma cells. Mol. Cell. Biol. 2000, 20, 6860–6871. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.; Faqeih, E.A.; Bartholdi, D.; Bole-Feysot, C.; Borochowitz, Z.; Cavalcanti, D.P.; Frigo, A.; Nitschke, P.; Roume, J.; Santos, H.G.; et al. Exome sequencing identifies inppl1 mutations as a cause of opsismodysplasia. Am. J. Hum. Genet. 2013, 92, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Funakoshi, A.; Miyasaka, K.; Iguchi, H.; Takiguchi, S. Sp-family of transcription factors regulates human SHIP2 gene expression. Gene 2005, 348, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Wu, T.T. Epigenetic alterations in gastric carcinogenesis. Cell Res. 2005, 15, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the “hallmarks of cancer”. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef] [PubMed]
- Black, A.R.; Black, J.D.; Azizkhan-Clifford, J. Sp1 and krüppel-like factor family of transcription factors in cell growth regulation and cancer. J. Cell Physiol. 2001, 188, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Davie, J.R. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann. Anat. 2010, 192, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Suske, G. The Sp-family of transcription factors. Gene 1999, 238, 291–300. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, C.K.; Oh, E.; Erkin, O.C.; Jung, H.S.; Cho, M.H.; Kwon, M.J.; Chae, S.W.; Kim, S.H.; Wang, L.H.; et al. Low SP1 expression differentially affects intestinal-type compared with diffuse-type gastric adenocarcinoma. PLoS ONE 2013, 8, e55522. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guan, X.; Gong, W.; Yao, J.; Peng, Z.; Wei, D.; Wu, T.T.; Huang, S.; Xie, K. Altered expression of transcription factor Sp1 critically impacts the angiogenic phenotype of human gastric cancer. Clin. Exp. Metastasis 2005, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wei, D.; Huang, S.; Peng, Z.; Le, X.; Wu, T.T.; Yao, J.; Ajani, J.; Xie, K. Transcription factor Sp1 expression is a significant predictor of survival in human gastric cancer. Clin. Cancer Res. 2003, 9, 6371–6380. [Google Scholar] [PubMed]
- Arakawa, N.; Sugai, T.; Habano, W.; Eizuka, M.; Sugimoto, R.; Akasaka, R.; Toya, Y.; Yamamoto, E.; Koeda, K.; Sasaki, A.; et al. Genome-wide analysis of DNA copy number alterations in early and advanced gastric cancers. Mol. Carcinog. 2016, 56, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xu, M.; Cui, X.; Liu, Y.; Zhang, Y.; Sui, Y.; Wang, D.; Peng, L.; Wang, D.; Yu, J. Aberrant expression of the candidate tumor suppressor gene DAL-1 due to hypermethylation in gastric cancer. Sci. Rep. 2016, 6, 21755. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liang, Q.; Li, X.; Tsoi, H.; Zhang, J.; Wang, H.; Go, M.Y.; Chiu, P.W.; Ng, E.K.; Sung, J.J.; et al. MDGA2 is a novel tumour suppressor cooperating with DMAP1 in gastric cancer and is associated with disease outcome. Gut 2016, 65, 1619–1631. [Google Scholar] [CrossRef] [PubMed]
- Hassler, M.R.; Klisaroska, A.; Kollmann, K.; Steiner, I.; Bilban, M.; Schiefer, A.I.; Sexl, V.; Egger, G. Antineoplastic activity of the DNA methyltransferase inhibitor 5-aza-2′-deoxycytidine in anaplastic large cell lymphoma. Biochimie 2012, 94, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I. Sp1: Emerging roles—Beyond constitutive activation of TATA-less housekeeping genes. Biochem. Biophys. Res. Commun. 2008, 372, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vizcaino, C.; Mansilla, S.; Portugal, J. Sp1 transcription factor: A long-standing target in cancer chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Colaluca, I.N.; Tosoni, D.; Nuciforo, P.; Senic-Matuglia, F.; Galimberti, V.; Viale, G.; Pece, S.; Di Fiore, P.P. Numb controls p53 tumour suppressor activity. Nature 2008, 451, 76–80. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Nucleotide Change | Status | Amino Acid Change | Location | Domain |
|---|---|---|---|---|---|
| HGC-27 | N | – | N | – | – |
| SGC-7901 | c.1134A>G | ho | p. 329Ser * | Ex9 | / |
| c.3395C>G | ho | p.Ala1083Gly | Ex26 | Pro-rich domain | |
| c.4046_4061del | ho | ? | Ex28 | / | |
| c.4192G>C | ho | ? | Ex28 | / | |
| MGC-803 | c.1143A>G | he | p. 329Ser * | Ex9 | / |
| c.C3395C>G | he | p.Ala1083Gly | Ex26 | Pro-rich domain | |
| c.4046_4061del | he | ? | Ex28 | / | |
| MKN-45 | c.1143A>G | ho | p. 329Ser * | Ex9 | / |
| c.3133C>A | he | p.Leu996Met | Ex26 | Pro-rich domain |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Y.; Qian, X.Y.; Xiao, M.M.; Shao, Y.L.; Guo, L.M.; Liao, D.P.; Da, J.; Zhang, L.J.; Xu, J. Decreased Sp1 Expression Mediates Downregulation of SHIP2 in Gastric Cancer Cells. Int. J. Mol. Sci. 2017, 18, 220. https://doi.org/10.3390/ijms18010220
Ye Y, Qian XY, Xiao MM, Shao YL, Guo LM, Liao DP, Da J, Zhang LJ, Xu J. Decreased Sp1 Expression Mediates Downregulation of SHIP2 in Gastric Cancer Cells. International Journal of Molecular Sciences. 2017; 18(1):220. https://doi.org/10.3390/ijms18010220
Chicago/Turabian StyleYe, Yan, Xue Yi Qian, Miao Miao Xiao, Yu Ling Shao, Li Mei Guo, Dong Ping Liao, Jie Da, Lin Jie Zhang, and Jiegou Xu. 2017. "Decreased Sp1 Expression Mediates Downregulation of SHIP2 in Gastric Cancer Cells" International Journal of Molecular Sciences 18, no. 1: 220. https://doi.org/10.3390/ijms18010220
APA StyleYe, Y., Qian, X. Y., Xiao, M. M., Shao, Y. L., Guo, L. M., Liao, D. P., Da, J., Zhang, L. J., & Xu, J. (2017). Decreased Sp1 Expression Mediates Downregulation of SHIP2 in Gastric Cancer Cells. International Journal of Molecular Sciences, 18(1), 220. https://doi.org/10.3390/ijms18010220