Experimental and Kinetic Study on Lignin Depolymerization in Water/Formic Acid System

Abstract
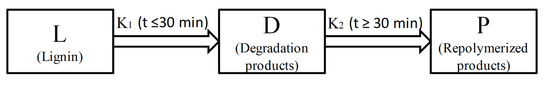
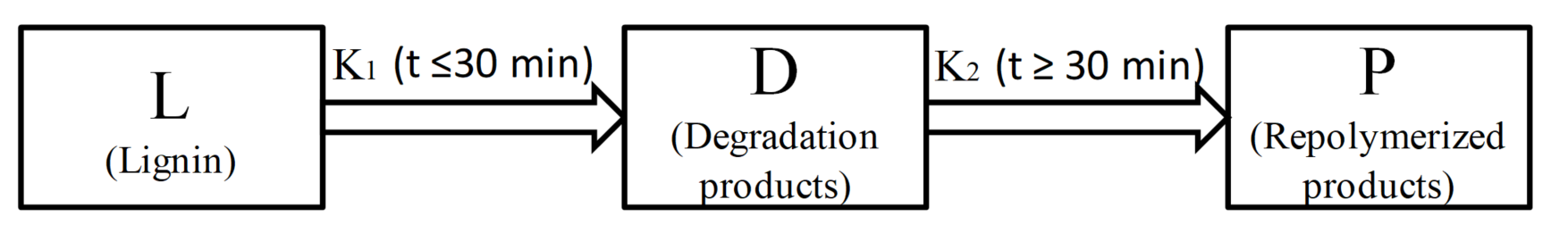
:
1. Introduction
2. Results and Discussion
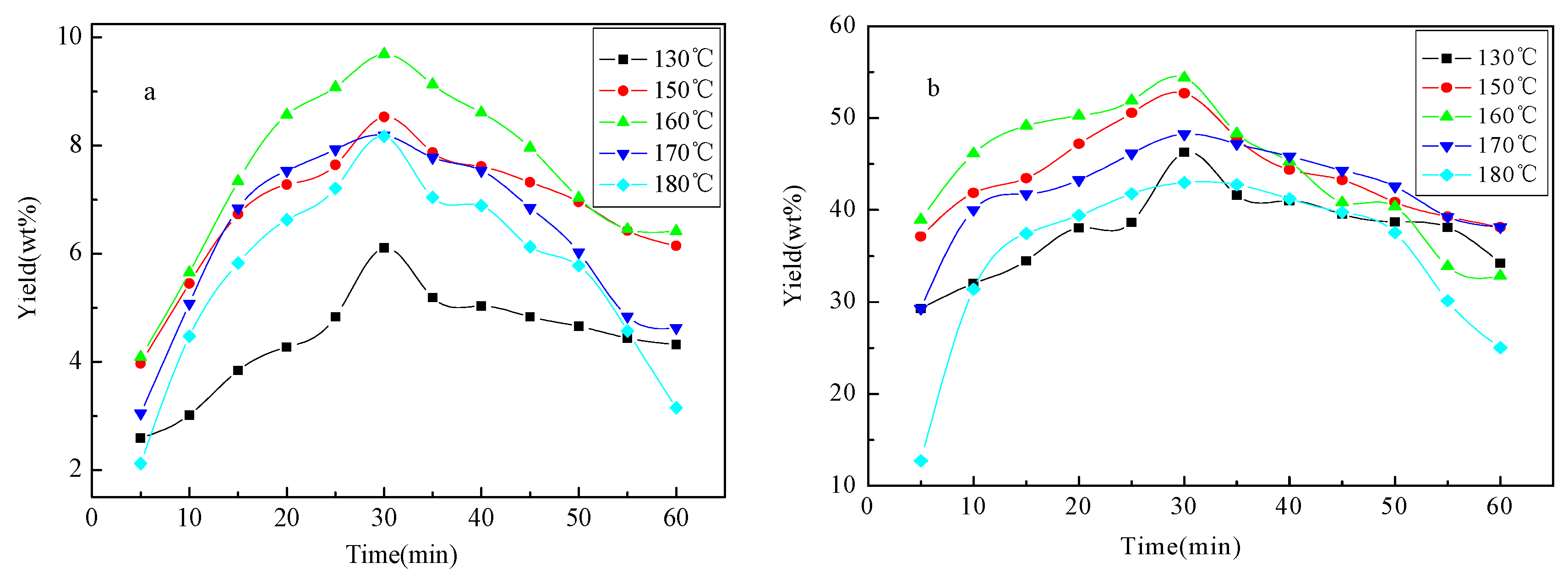
2.1. Yield of Liquid Products
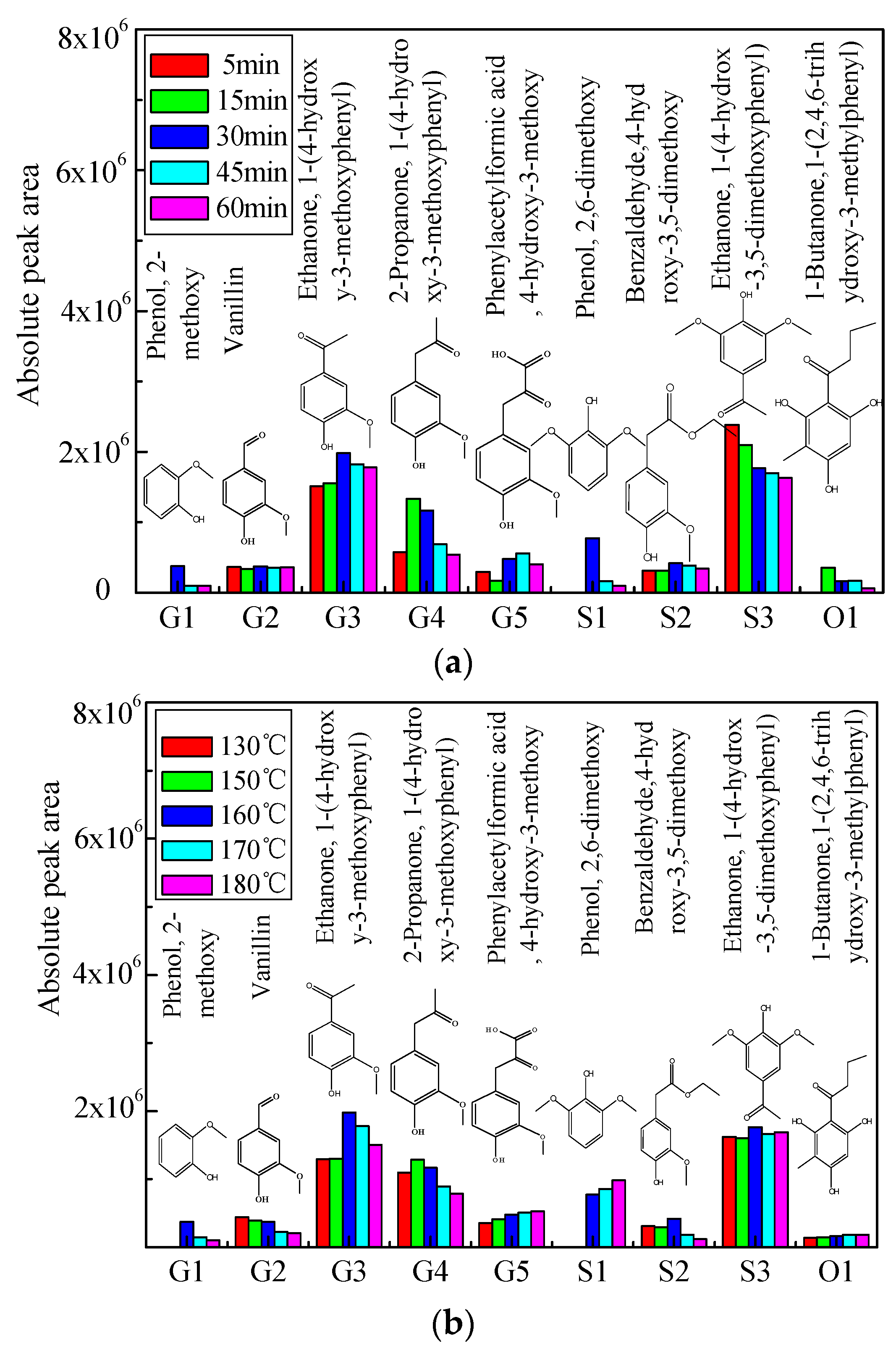
2.2. Distribution of Aromatic Monomers in Bio-Oil 1
2.3. Characterization of Aromatic Oligomers in Bio-Oil 2
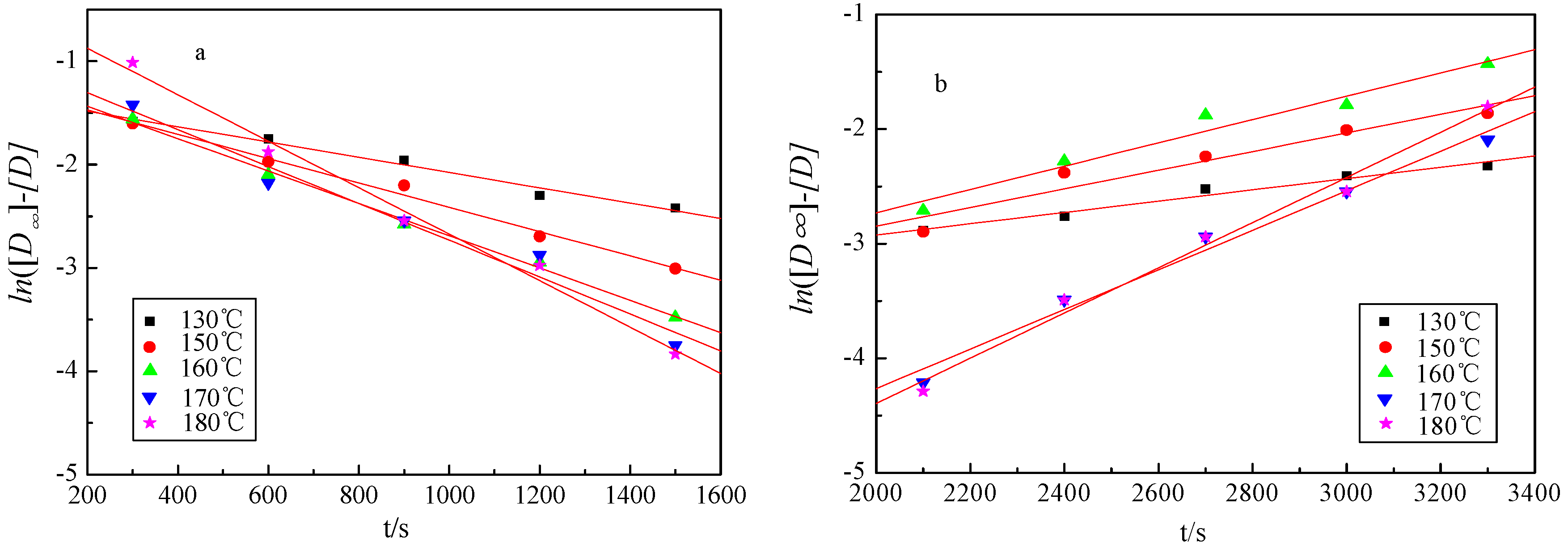
2.4. Kinetics of Microwave-Assisted Solvolysis of Lignin
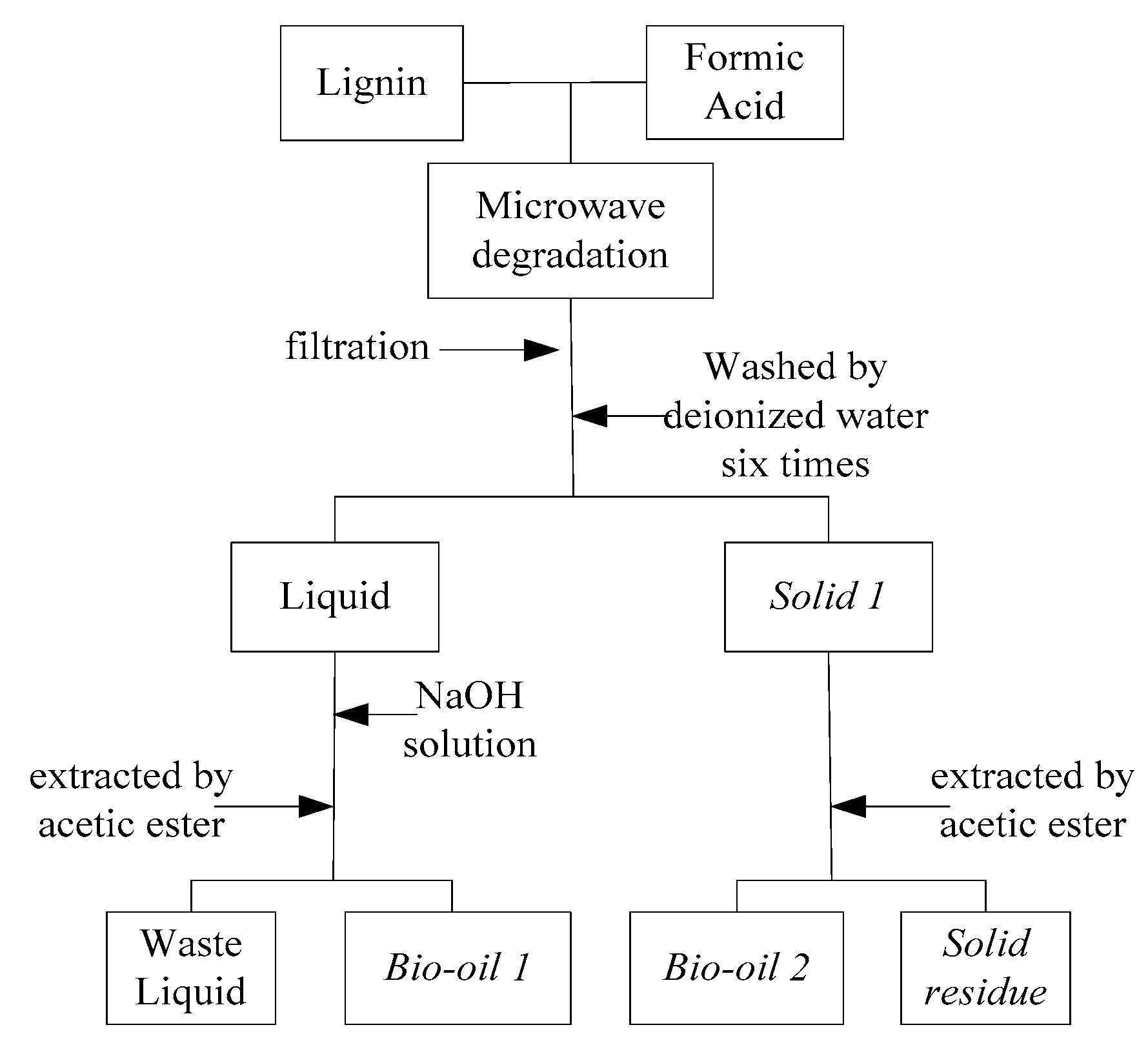
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McKendry, P. Energy production from biomass (part 1): Overview of biomass. Bioresour. Technol. 2002, 83, 37–46. [Google Scholar] [CrossRef]
- Hu, J.; Shen, D.; Xiao, R.; Wu, S.; Zhang, H. Free-Radical Analysis on Thermochemical Transformation of Lignin to Phenolic Compounds. Energy Fuels 2012, 27, 285–293. [Google Scholar] [CrossRef]
- Calvo-Flores, F.G.; Dobado, J.A. Lignin as renewable raw material. ChemSusChem 2010, 3, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Beall, F.C. Thermogravimetric analysis of wood lignin and hemicelluloses. Wood Fiber Sci. 1969, 3, 215–226. [Google Scholar]
- Wahyudiono, M.S.; Goto, M. Decomposition of lignin alkaline and chemicals recovery in sub-and supercritical water. In Proceedings of the ISASF (International Society for Advancement of Supercritical Fluids), Trieste, Italy, 13–16 June 2004. [Google Scholar]
- Kleinert, M.; Barth, T. Phenols from lignin. Chem. Eng. Technol. 2008, 31, 736–745. [Google Scholar] [CrossRef]
- Gosselink, R.J.; Teunissen, W.; van Dam, J.E.; de Jong, E.; Gellerstedt, G.; Scott, E.L.; Sanders, J.P. Lignin depolymerisation in supercritical carbon dioxide/acetone/water fluid for the production of aromatic chemicals. Bioresour. Technol. 2011, 106, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Goto, M. Recovery of phenolic compounds through the decomposition of lignin in near and supercritical water. Chem. Eng. Process. 2008, 47, 1609–1619. [Google Scholar]
- Pińkowska, H.; Wolak, P.; Złocińska, A. Hydrothermal decomposition of alkali lignin in sub-and supercritical water. Chem. Eng. J. 2012, 187, 410–414. [Google Scholar] [CrossRef]
- Ye, Y.; Zhang, Y.; Fan, J.; Chang, J. Novel method for production of phenolics by combining lignin extraction with lignin depolymerization in aqueous ethanol. Ind. Eng. Chem. Res. 2011, 51, 103–110. [Google Scholar] [CrossRef]
- Xiao, W.; Han, L.; Zhao, Y. Comparative study of conventional and microwave-assisted liquefaction of corn stover in ethylene glycol. Ind. Crops Prod. 2011, 34, 1602–1606. [Google Scholar] [CrossRef]
- Bu, Q.; Lei, H.; Ren, S.; Wang, L.; Zhang, Q.; Tang, J.; Ruan, R. Production of phenols and biofuels by catalytic microwave pyrolysis of lignocellulosic biomass. Bioresour. Technol. 2012, 108, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Badamali, S.K.; Clark, J.H.; Breeden, S.W. Microwave assisted selective oxidation of lignin model phenolic monomer over SBA-15. Catal. Commun. 2008, 9, 2168–2170. [Google Scholar] [CrossRef]
- Toledano, A.; Serrano, L.; Labidi, J.; Pineda, A.; Balu, A.M.; Luque, R. Heterogeneously Catalysed Mild Hydrogenolytic Depolymerisation of Lignin Under Microwave Irradiation with Hydrogen-Donating Solvents. ChemCatChem 2013, 5, 977–985. [Google Scholar] [CrossRef]
- Rahimi, A.; Ulbrich, A.; Coon, J.J.; Stahl, S.S. Formic-acid-induced depolymerization of oxidized lignin to aromatics. Nature 2014, 515, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Dang, Y.; Song, C.; Guo, J.; Wang, Z.X. Depolymerization of Oxidized Lignin Catalyzed by Formic Acid Exploits an Unconventional Elimination Mechanism Involving 3c–4e Bonding: A DFT Mechanistic Study. ACS. Catal. 2015, 5, 6386–6396. [Google Scholar] [CrossRef]
- Zhang, B.; Huang, H.-J.; Ramaswamy, S. Reaction kinetics of the hydrothermal treatment of lignin. Appl. Biochem. Biotechnol. 2008, 147, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zaixiong, L. Study on Oxidation of Alkali Lignin Assisted by Microwave Irradiation. Master’s Thesis, South China University of Technology, Guangzhou, China, 2011. [Google Scholar]
- Hu, J.; Xiao, R.; Shen, D.; Zhang, H. Structural analysis of lignin residue from black liquor and its thermal performance in thermogravimetric-Fourier transform infrared spectroscopy. Bioresour. Technol. 2012, 128, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Nowakowski, D.J.; Bridgwater, A.V. Effect of the temperature on the composition of lignin pyrolysis products. Energy Fuels 2010, 24, 4470–4475. [Google Scholar] [CrossRef]
- Del Río, J.C.; Rencoret, J.; Gutiérrez, A.; Nieto, L.; Jiménez-Barbero, J.; Martínez, Á.T. Structural characterization of guaiacyl-rich lignins in flax (Linum usitatissimum) fibers and shives. J. Agric. Food Chem. 2011, 59, 11088–11099. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Molecular Weight a | Peak (m/z) | Relative Peak Area b (%) | ||||
|---|---|---|---|---|---|---|---|
| 5 min | 15 min | 30 min | 45 min | 60 min | |||
| i | 274 | 272–274 | 8.0 | 7.5 | 8.6 | 8.1 | 8.0 |
| ii | 290 | 283–294 | 0 | 5.0 | 5.7 | 3.5 | 3.0 |
| iii | 318 | 311–318 | 4.5 | 7.3 | 8.4 | 8.6 | 9.1 |
| iv | 328 | 319–328 | 11.3 | 11.6 | 16.4 | 12.5 | 8.2 |
| v | 342 | 330–342 | 6.3 | 6.4 | 6.1 | 6.7 | 6.1 |
| vi | 358 | 350–358 | 13.9 | 8.5 | 9.5 | 8.6 | 9.7 |
| vii | 378 | 368–378 | 4.0 | 3.8 | 2.5 | 1.8 | 2.6 |
| viii | 394 | 384–394 | 14.5 | 11.5 | 10.9 | 6.5 | 7.0 |
| ix | 424 | 424 | 8.9 | 5.9 | 4.2 | 5.0 | 5.5 |
| Total | 71.4 | 67.5 | 72.3 | 61.3 | 59.2 | ||
| No. | Molecular Weight a | Peak (m/z) | Relative Peak Area b (%) | ||||
|---|---|---|---|---|---|---|---|
| 130 °C | 150 °C | 160 °C | 170 °C | 180 °C | |||
| i | 274 | 272–274 | 9.4 | 8.9 | 8.6 | 8.3 | 10.4 |
| ii | 290 | 283–294 | 4.5 | 7.5 | 5.7 | 4.3 | 3.4 |
| iii | 318 | 311–318 | 7.9 | 7.9 | 8.4 | 7.0 | 6.4 |
| iv | 328 | 319–328 | 19.3 | 18.8 | 16.4 | 11.1 | 1.6 |
| v | 342 | 330–342 | 9.4 | 9.2 | 6.1 | 7.1 | 9.8 |
| vi | 358 | 350–358 | 11.5 | 6.8 | 9.5 | 10.5 | 14.8 |
| vii | 378 | 368–378 | 5.1 | 4.8 | 2.5 | 4.2 | 11.9 |
| viii | 394 | 384–394 | 5.9 | 6.0 | 10.9 | 13.8 | 19.4 |
| ix | 424 | 424 | 2.4 | 3.1 | 4.2 | 10.2 | 8.0 |
| Total | 75.4 | 73 | 72.3 | 76.5 | 85.7 | ||
| Temperature (°C) | The First Stage | The Second Stage | ||
|---|---|---|---|---|
| k (L·mol−1·s−1) | R | k (L·mol−1·s−1) | R | |
| 130 | 0.000739 | 0.99036 | 0.000493 | 0.98531 |
| 150 | 0.00118 | 0.99535 | 0.000814 | 0.9655 |
| 160 | 0.00156 | 0.99823 | 0.00102 | 0.98169 |
| 170 | 0.00179 | 0.98472 | 0.00173 | 0.99257 |
| 180 | 0.00225 | 0.99453 | 0.00197 | 0.99391 |
| Reaction | The Linear Fitting Equation | R | Ea (kJ·mol−1) | A (L·mol−1·s−1) |
|---|---|---|---|---|
| The first stage | lnK = 2.8430 − 4049.35/T | 0.99792 | 33.67 | 17.1672 |
| The second stage | lnK = 5.4203 − 5276.81/T | 0.98374 | 43.87 | 225.9469 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Guan, S.; Shen, D. Experimental and Kinetic Study on Lignin Depolymerization in Water/Formic Acid System. Int. J. Mol. Sci. 2017, 18, 2082. https://doi.org/10.3390/ijms18102082
Wang Q, Guan S, Shen D. Experimental and Kinetic Study on Lignin Depolymerization in Water/Formic Acid System. International Journal of Molecular Sciences. 2017; 18(10):2082. https://doi.org/10.3390/ijms18102082
Chicago/Turabian StyleWang, Qi, Sipian Guan, and Dekui Shen. 2017. "Experimental and Kinetic Study on Lignin Depolymerization in Water/Formic Acid System" International Journal of Molecular Sciences 18, no. 10: 2082. https://doi.org/10.3390/ijms18102082
APA StyleWang, Q., Guan, S., & Shen, D. (2017). Experimental and Kinetic Study on Lignin Depolymerization in Water/Formic Acid System. International Journal of Molecular Sciences, 18(10), 2082. https://doi.org/10.3390/ijms18102082