Chemokines from a Structural Perspective



Abstract
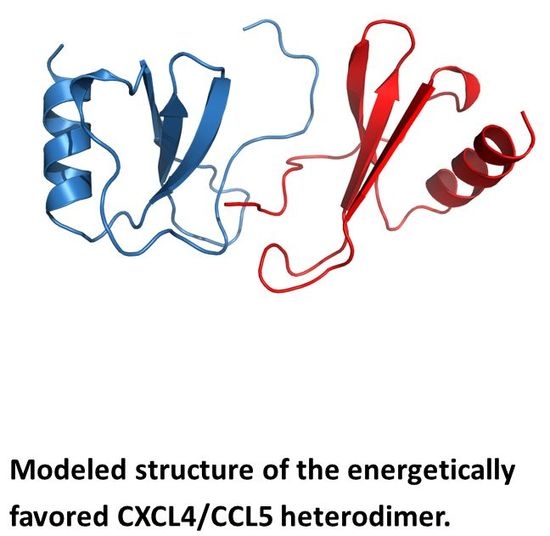
:
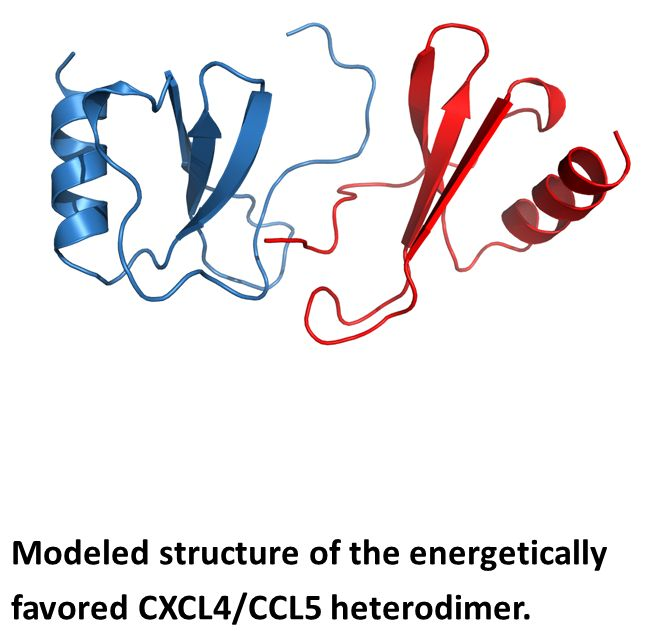{kind=link}
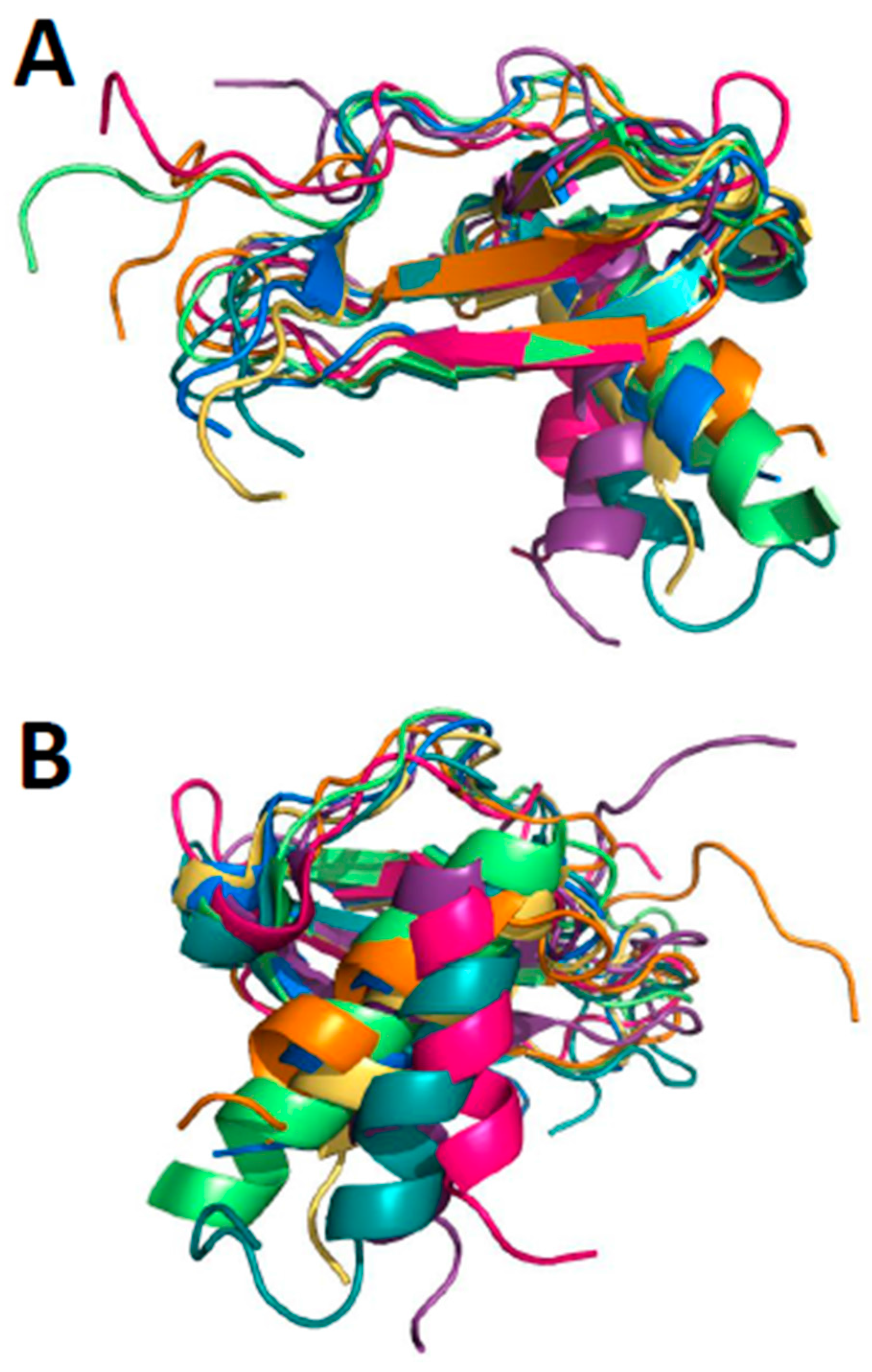{kind=link}
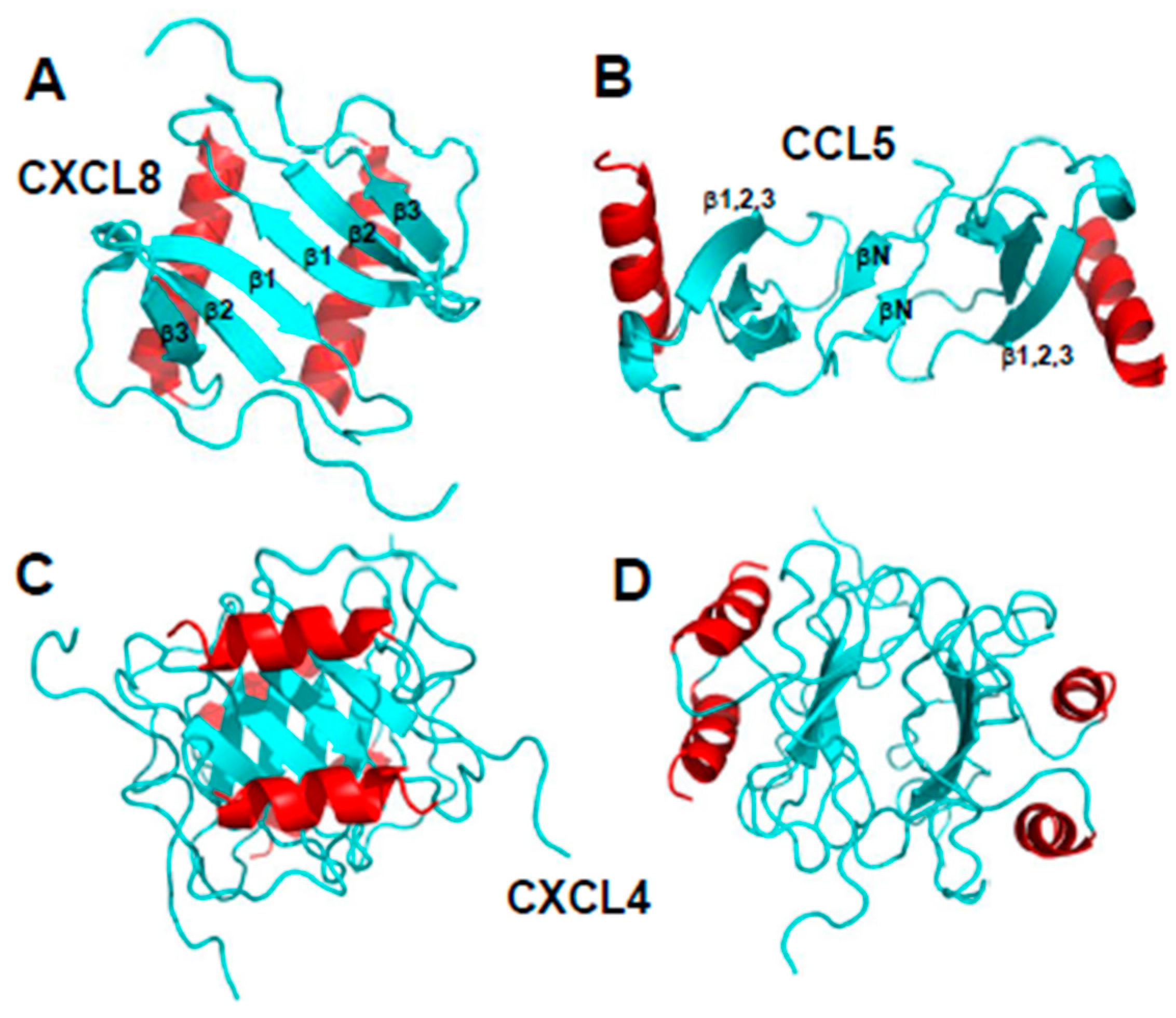{kind=link}
{kind=link}
{kind=link}
1. Chemokine Structures
2. Chemokine Heterodimers
3. Functional Impact of Chemokine Structure
4. Chemokine Antagonists
Note
Conflicts of Interest
References
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Mackay, C.R. Chemokines:Immunology’s high impact factors. Nat. Immunol. 2001, 2, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Youn, B.S.; Mantel, C.; Broxmeyer, H.E. Chemokines, chemokine receptors and hematopoiesis. Immunol. Rev. 2000, 177, 150–174. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Elner, S.G.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258, 1798–1801. [Google Scholar] [CrossRef]
- Belperio, J.A.; Keane, M.P.; Arenberg, D.A.; Addison, C.L.; Ehlert, J.E.; Burdick, M.D.; Strieter, R.M. CXC Chemokines in Angiogenesis. J. Leukoc. Biol. 2000, 68, 1–8. [Google Scholar] [PubMed]
- Zlotnik, A.; Yoshie, O. Chemokines: A new classification system and their role in immunity. Immunity 2000, 12, 121–127. [Google Scholar] [CrossRef]
- Clore, G.M.; Gronenborn, A.M. Three-dimensional structures of alpha and beta chemokines. FASEB J. 1995, 9, 57–62. [Google Scholar] [PubMed]
- St. Charles, R.; Walz, D.A.; Edwards, B.F. Crystal structure of bovine platelet factor-4. J. Biol. Chem. 1989, 264, 2092–2099. [Google Scholar]
- Mayo, K.H.; Yang, Y.; Daly, T.J.; Barry, J.K.; La Rosa, G.J. Secondary Structure of Neutrophil Activating Peptide-2 Determined by 1H-NMR Spectroscopy. Biochem. J. 1994, 303, 371–376. [Google Scholar] [CrossRef]
- Clore, G.M.; Appella, E.; Yamada, M.; Matsushima, K.; Gronenborn, A.M. Three dimensional structure of interleukin 8 in solution. Biochemistry 1990, 29, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- Handel, T.M.; Domaille, P.J. Heteronuclear (1H, 13C, 15N) NMR assignments and solution structure of the monocyte chemoattractant protein-1 (MCP-1) dimer. Biochemistry 1996, 35, 6569–6584. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.A.; Buelow, B.J.; Nevins, A.M.; Jones, S.E.; Peterson, F.C.; Gundry, R.L.; Grayson, M.H.; Volkman, B.F. Structure-function analysis of CCL28 in the development of post-viral asthma. J. Biol. Chem. 2015, 290, 4528–4536. [Google Scholar] [CrossRef] [PubMed]
- Mayo, K.H.; Chen, M.-J. Human Platelet Factor 4 Monomer- Dimer-Tetramer Equilibria Investigated by NMR Spectroscopy. Biochemistry 1989, 28, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Clark-Lewis, I.; Kim, K.S.; Rajarathnam, K.; Gong, J.H.; Dewald, B.; Moser, B.; Baggiolini, M.; Sykes, B.D. Structure-activity relationships of chemokines. J. Leukoc. Biol. 1995, 57, 703–711. [Google Scholar] [PubMed]
- Wang, X.; Watson, C.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Oligomeric structure of the chemokine CCL5/RANTES from NMR, MS, and SAXS data. Structure 2011, 19, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Jansma, A.L.; Kirkpatrick, J.P.; Hsu, A.R.; Handel, T.M.; Nietlispach, D. NMR analysis of the structure, dynamics, and unique oligomerization properties of the chemokine CCL27. J. Biol. Chem. 2010, 285, 14424–14437. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.G.; Triandafillou, C.G.; Huang, T.Y.; Zulueta, M.M.; Banerjee, S.; Dinner, A.R.; Hung, S.C.; Tang, W.J. Crystal structure of CC chemokine 5 (CCL5). Proc. Natl. Acad. Sci. USA 2016, 113, 5000–5005. [Google Scholar] [CrossRef] [PubMed]
- Mayo, K.H.; Roongta, V.; Barker, S.; Milius, R.; Ilyina, E.; Quinlan, C.; La Rosa, G.; Daly, T. NMR Solution Structure of the 32 kD Tetrameric Platelet Factor-4 ELR-Motif N-terminal Chimer: A Symmetric Tetramer. Biochemistry 1995, 34, 11399–11409. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mayo, K.H.; Daly, T.; Barry, J.K.; La Rosa, G.J. Subunit Association and Structural Analysis of Platelet Basic Protein and Related Proteins Investigated by 1H-NMR Spectroscopy and Circular Dichroism. J. Biol. Chem. 1994, 269, 20110–20118. [Google Scholar] [PubMed]
- Swaminathan, G.J.; Holloway, D.E.; Colvin, R.A.; Campanella, G.K.; Papageorgiou, A.C.; Luster, A.D.; Acharya, K.R. Crystal Structures of Oligomeric Forms of the IP-10/CXCL10 Chemokine. Structure 2003, 11, 521–532. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.; Bancroft, D.P.; Lai, C.K.; Maione, T.E. Crystal structure of recombinant human platelet factor 4. Biochemistry 1994, 33, 8361–8366. [Google Scholar] [CrossRef] [PubMed]
- Lubkowski, J.; Bujacz, G.; Boqué, L.; Domaille, P.J.; Handel, T.M.; Wlodawer, A. The structure of MCP-1 in two crystal forms provides a rare example of variable quaternary interactions. Nat. Struct. Biol. 1997, 4, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, D.V.; Young, H.; Linhardt, R.J.; Mayo, K.H. Heparin Dodecasaccharide Binding to Platelet Factor-4 and Growth-related Protein-α: Induction of a Partially Folded State and Implications for Heparin-Induced Thrombocytopenia. J. Biol. Chem. 1999, 274, 25317–25329. [Google Scholar] [CrossRef] [PubMed]
- Jansma, A.; Handel, T.M.; Hamel, D.J. Chapter 2. Homo- and hetero-oligomerization of chemokines. Methods Enzymol. 2009, 461, 31–50. [Google Scholar] [PubMed]
- Paoletti, S.; Petkovic, V.; Sebastiani, S.; Danelon, M.G.; Uguccioni, M.; Gerber, B.O. A rich chemokine environment strongly enhances leukocyte migration and activities. Blood 2005, 105, 3405–3412. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.S.; Grimm, J.; Manice, L.A.; Colvin, R.A.; Medoff, B.D.; Wojtkiewicz, G.R.; Weissleder, R.; Luster, A.D. Oligomerization of CXCL10 Is Necessary for Endothelial Cell Presentation and In Vivo Activity. J. Immunol. 2006, 177, 6991–6998. [Google Scholar] [CrossRef] [PubMed]
- Rek, A.; Brandner, B.; Geretti, E.; Kungl, A.J. A biophysical insight into the RANTES–glycosamino- glycan interaction. Biochim. Biophys. Acta 2009, 1794, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Guo, Q.; Guo, L.; Lenz, M.; Qian, F.; Koenen, R.R.; Xu, H.; Schilling, A.B.; Weber, C.; Ye, R.D.; et al. Polymerization of MIP-1 chemokine (CCL3 and CCL4) and clearance of MIP1 by insulin-degrading enzyme. EMBO J. 2010, 29, 3952–3966. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.J.; Mayo, K.H. Human Platelet Factor 4 Subunit Association-Dissociation Thermodynamics and Kinetics. Biochemistry 1991, 30, 6402–6411. [Google Scholar] [CrossRef] [PubMed]
- Mayo, K.H. Low Affinity Platelet Factor 4 1H-NMR Derived Aggregate Equilibria Indicate Physiological Preference for Monomers over Dimers and Tetramers. Biochemistry 1991, 30, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Young, H.; Roongta, V.; Daly, T.J.; Mayo, K.H. NMR Structure and Dynamics of Monomeric Neutrophil Activating Peptide-2. Biochem. J. 1999, 338, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Nesmelova, I.V.; Sham, Y.; Gao, J.; Mayo, K.H. CXC-chemokines associate with CC-chemokines to form mixed heterodimers: Rantes and PF4 monomers associate as CC-type heterodimers. J. Biol. Chem. 2008, 283, 24155–24166. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mayo, K.H. Alcohol-Induced Protein Folding Transitions in Platelet Factor- 4: The O-State. Biochemistry 1993, 32, 8661–8671. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Barker, S.; Chen, M.-J.; Mayo, K.H. Effect of Low Molecular Weight Aliphatic Alcohols and Related Compounds on Platelet Factor-4 Subunit Association. J. Biol. Chem. 1993, 268, 9223–9229. [Google Scholar] [PubMed]
- Veldkamp, C.T.; Ziarek, J.J.; Su, J.; Basnet, H.; Lennertz, R.; Weiner, J.J.; Peterson, F.C.; Baker, J.E.; Volkman, B.F. Monomeric structure of the cardio-protective chemokine SDF-1/CXCL12. Protein Sci. 2009, 18, 1359–1369. [Google Scholar] [CrossRef] [PubMed]
- Veldkamp, C.T.; Peterson, F.C.; Pelzek, A.J.; Volkman, B.F. The monomer-dimer equilibrium of stromal cell-derived factor-1 (CXCL12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005, 14, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Rajarathnam, K.; Kim, K.-S.; Clark-Lewis, I.; Sykes, B.D. Solution Structure of Eotaxin, a Chemokine That Selectively Recruits Eosinophils in Allergic Inflammation. J. Biol. Chem. 1998, 273, 22471–22479. [Google Scholar] [CrossRef] [PubMed]
- Guan, E.; Wang, J.; Norcross, M.A. Identification of Human Macrophage Inflammatory Proteins 1alpha and 1beta as a Native Secreted Heterodimer. J. Biol. Chem. 2001, 276, 12404–12409. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.Z.; Nesmelova, I.; Mayo, K.H.; Verfaillie, C.M.; Pitchford, E.; Slungaard, A. Platelet Factor 4 Promotes Adhesion of Hematopoietic Progenitor Cells and Binds IL-8: Novel Mechanisms for Modulation of Hematopoiesis. Blood 2003, 101, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Nesmelova, I.; Sham, Y.; Dudek, A.Z.; van Eijk, L.I.; Wu, G.; Slungaard, A.; Mortari, F.; Griffioen, A.W.; Mayo, K.H. Platelet Factor 4 and Interleukin-8 CXC Chemokine Heterodimer Formation Modulates Function at the Quaternary Structural Level. J. Biol. Chem. 2005, 280, 4948–4958. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.; von Hundelhausen, P.; Nesmelova, I.V.; Zernecke, A.; Liehn, E.A.; Sarabi, A.; Kramp, B.K.; Piccinini, A.; Paludan, S.R.; Kowalska, M.A.; et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat. Med. 2009, 15, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Agten, S.; Eckardt, V.; Schmitt, M.; Blanchet, X.; Neideck, C.; Ippel, H.; Bidzhekov, K.; Wichapong, K.; Faussner, A.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9, 384. [Google Scholar] [CrossRef] [PubMed]
- Ellyard, J.I.; Simson, L.; Bezos, A.; Johnston, K.; Freeman, C.; Parish, C.R. Eotaxin Selectively Binds Heparin. An interaction that protects eotaxin from proteolysis and potentiates chemotactic activity in vivo. J. Biol. Chem. 2007, 282, 15238–15247. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E. The BBXB Motif of RANTES Is the Principal Site for Heparin Binding and Controls Receptor Selectivity. J. Biol. Chem. 2001, 276, 10620–10626. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.J.; Oh, Y.I.; Chang, S.-K.; Hsieh-Wilson, L.C. Tunable Heparan Sulfate Mimetics for Modulating Chemokine Activity. J. Am. Chem. Soc. 2013, 135, 10898–10901. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Yarovoi, S.V.; Zhu, Z.; Rauova, L.; Hayes, V.; Lebedeva, T.; Liu, Q.; Poncz, M.; Arepally, G.; Cines, D.B.; et al. Crystal structure of platelet factor 4 complexed with fondaparinux. Nat. Commun. 2015, 6, 8277. [Google Scholar] [CrossRef] [PubMed]
- Salanga, C.L.; Handel, T.M. Chemokine oligomerization and interactions with receptors and glycosaminoglycans: The role of structural dynamics in function. Exp. Cell Res. 2011, 317, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E. Chemokines and Glycosaminoglycans. Front. Immunol. 2015, 6, 246. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.P.; Johnson, Z.; Borlat, F.; Zwahlen, C.; Kungl, A.; Roulin, K.; Harrenga, A.; Wells, T.N.; Proudfoot, A.E. The X-Ray Structure of RANTES Heparin-Derived Disaccharides Allows the Rational Design of Chemokine Inhibitors. Structure 2004, 12, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.; Andaya, A.; Bleiholder, C.; Leary, J.A. Differentiation of CC vs. CXC Chemokine Dimers with GAG Octasaccharide Binding Partners: An Ion Mobility Mass Spectrometry Approach. J. Am. Chem. Soc. 2013, 135, 4325–4332. [Google Scholar] [CrossRef] [PubMed]
- Mayo, K.H.; Ilyina, E.; Roongta, V.; Dundas, M.; Joseph, J.; Lai, C.K.; Maione, T.; Daly, T. Heparin Binding to Platelet Factor-4. An NMR and Site-Directed Mutagenesis Study: Arginine Residues Crucial for Binding. Biochem. J. 1995, 312, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.C.; Tyler, R.C.; Peterson, F.C.; Dyer, D.P.; Zhang, F.; Linhardt, R.J.; Handel, T.M.; Volkman, B.F. Examination of Glycosaminoglycan Binding Sites on the XCL1 Dimer. Biochemistry 2016, 55, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Hoogewerf, A.J.; Kuschert, G.S.; Proudfoot, A.E.; Borlat, F.; Clark-Lewis, I.; Power, C.A.; Wells, T.N. Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 1997, 36, 13570–13578. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The dependence of chemokine-glycosaminoglycan interactions on chemokine oligomerization. Glycobiology 2016, 26, 312–326. [Google Scholar] [CrossRef] [PubMed]
- Crown, S.E.; Yu, Y.; Sweeney, M.D.; Leary, J.A.; Handel, T.M. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem. 2006, 281, 25438–25446. [Google Scholar] [CrossRef] [PubMed]
- Verkaar, F.; van Offenbeek, J.; van der Lee, M.M.C.; van Lith, L.H.C.J.; Watts, A.O.; Rops, A.L.W.M.M.; Aguilar, D.C.; Ziarek, J.J.; van der Vlag, J.; Handel, T.M.; et al. Chemokine cooperativity is caused by competitive glycosaminoglycan binding. J. Immunol. 2014, 192, 3908–3914. [Google Scholar] [CrossRef] [PubMed]
- Koenen, R.R.; Weber, C. Therapeutic targeting of chemokine interactions in atherosclerosis. Nat. Rev. Drug Discov. 2010, 9, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Viola, A.; Luster, A.D. Chemokines and Their Receptors: Drug Targets in Immunity and Inflammation. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 171–197. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.J.; Crown, S.E.; Handel, T.M. Chemokine: Receptor structure, interactions, and antagonism. Annu. Rev. Immunol. 2007, 25, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Das, B.B.; Casagrande, F.; Tian, Y.; Nothnagel, H.J.; Chu, M.; Kiefer, H.; Maier, K.; De Angelis, A.A.; Marassi, F.M.; Opella, S.J. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature 2012, 491, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Thelen, M.; Stein, J.V. How chemokines invite leukocytes to dance. Nat. Immunol. 2008, 9, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Chien, E.Y.; Mol, C.D.; Fenalti, G.; Liu, W.; Katritch, V.; Abagyan, R.; Brooun, A.; Wells, P.; Bi, F.C.; et al. Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Van Raemdonck, K.; Van den Steen, P.E.; Liekens, S.; Van Damme, J.; Struyf, S. CXCR3 ligands in disease and therapy. Cytokine Growth Factor Rev. 2015, 26, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, L.; Rajarathnam, K. Structural Basis of Chemokine Receptor Function—A Model for Binding Affinity and Ligand Selectivity. Biosci. Rep. 2006, 26, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Katancik, J.A.; Sharma, A.; Radel, S.J.; De Nardin, E. Mapping of the extracellular binding regions of the human interleukin-8 type B receptor. Biochem. Biophys. Res. Commun. 1997, 232, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Skelton, N.J.; Quan, C.; Reilly, D.; Lowman, H. Structure of a CXC chemokine-receptor fragment in complex with interleukin-8. Struct. Fold. Des. 1999, 7, 157–168. [Google Scholar] [CrossRef]
- Handel, T.M.; Lau, E.K. Chemokine structure and receptor interactions. Ernst Scher Res. Found. Workshop 2004, 2004, 101–124. [Google Scholar]
- Casarosa, P.; Waldhoer, M.; LiWang, P.J.; Vischer, H.F.; Kledal, T.; Timmerman, H.; Schwartz, T.W.; Smit, M.J.; Leurs, R. CC and CX3C chemokines differentially interact with the N terminus of the human cytomegalovirus-encoded US28 receptor. J. Biol. Chem. 2005, 280, 3275–3285. [Google Scholar] [CrossRef] [PubMed]
- Hemmerich, S.; Paavola, C.; Bloom, A.; Bhakta, S.; Freedman, R.; Grunberger, D.; Krstenansky, J.; Lee, S.; McCarley, D.; Mulkins, M.; et al. Identification of residues in the mono cytechemotacticprotein-1 that contact the MCP-1 receptor, CCR2. Biochemistry 1999, 38, 13013–13025. [Google Scholar] [CrossRef] [PubMed]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Dzuiba, J.; Van Damme, J.; Walz, A.; Marriott, D.; et al. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef] [PubMed]
- Blanpain, C.; Doranz, B.J.; Bondue, A.; Govaerts, C.; De Leener, A.; Vassart, G.; Doms, R.W.; Proudfoot, A.; Parmentier, M. The core domain of chemokines bind CCR5 extracellular domains while their amino terminus interacts with the transmembrane helix bundle. J. Biol. Chem. 2003, 278, 5179–5187. [Google Scholar] [CrossRef] [PubMed]
- Crump, M.P.; Gong, J.H.; Loetscher, P.; Rajarathnam, K.; Amara, A.; Arenzana-Seisdedos, F.; Virelizier, J.L.; Baggiolini, M.; Sykes, B.D.; Clark-Lewis, I. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 1997, 16, 6996–7007. [Google Scholar] [CrossRef] [PubMed]
- Campanella, G.S.; Lee, E.M.; Sun, J.; Luster, A.D. CXCR3 and heparin binding sites of the chemokine IP-10 (CXCL10). J. Biol. Chem. 2003, 278, 17066–17074. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Kufareva, I.; Holden, L.G.; Wang, C.; Zheng, Y.; Zhao, C.; Fenalti, G.; Wu, H.; Han, G.W.; Cherezov, V.; et al. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science 2015, 357, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Handel, T.M. The structure of a CXCR4-chemokine complex. Front. Immunol. 2015, 6, 282. [Google Scholar] [CrossRef] [PubMed]
- Ziarek, J.J.; Kleist, A.B.; London, N.; Raveh, B.; Montpas, N.; Bonneterre, J.; St-Onge, G.; DiCosmo-Ponticello, C.J.; Koplinski, C.A.; Roy, I.; et al. Structural basis for chemokine recognition by a G protein-coupled receptor and implications for receptor activation. Sci. Signal. 2017, 10, 471. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Gustavsson, M.; Holden, L.G.; Qin, L.; Zheng, Y.; Handel, T.M. Disulfide trapping for modeling and structure determination of receptor:chemokine complexes. Methods Enzymol. 2016, 570, 389–420. [Google Scholar] [PubMed]
- Veldkamp, C.T.; Seibert, C.; Peterson, F.C.; De la Cruz, N.B.; Haugner, J.C.; Basnet, H.; Sakmar, T.P.; Volkman, B.F. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci. Signal. 2008, 1, ra4. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Stephens, B.S.; Holden, L.G.; Qin, L.; Zhao, C.; Kawamura, T.; Abagyan, R.; Handel, T.M. Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: Molecular modeling and experimental validation. Proc. Natl. Acad. Sci. USA 2014, 111, 5363–5372. [Google Scholar] [CrossRef] [PubMed]
- Kleist, A.B.; Getschman, A.E.; Ziarek, J.J.; Nevins, A.M.; Gauthier, P.A.; Chevigné, A.; Szpakowska, M.; Volkman, B.F. New paradigms in chemokine receptor signal transduction: Moving beyond the two-site model. Biochem. Pharmacol. 2016, 114, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Drury, L.J.; Ziarek, J.J.; Gravel, S.; Veldkamp, C.T.; Takekoshi, T.; Hwang, S.T.; Heveker, N.; Volkman, B.F.; Dwinell, M.B. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 17655–17660. [Google Scholar] [CrossRef] [PubMed]
- Hensbergen, P.J.; Wijnands, P.G.J.; Schreurs, M.W.J.; Scheper, R.J.; Willemze, R.; Tensen, C.P. The CXCR3 Targeting Chemokine CXCL11 Has Potent Antitumor Activity In Vivo Involving Attraction of CD8+ T Lymphocytes But Not Inhibition of Angiogenesis. J. Immunother. 2005, 28, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Uguccioni, M. Modulation of Chemokine Responses: Synergy and Cooperativity. Front. Immunol. 2016, 7, 183. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M. Synergy between proinflammatory ligands of G protein-coupled receptors in neutrophil activation and migration. J. Leukoc. Biol. 2004, 76, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Struyf, S.; Noppen, S.; Schutyser, E.; Springael, J.Y.; Parmentier, M.; Proost, P.; Van Damme, J. Synergy between Co-produced CC and CXC Chemokines in Monocyte Chemotaxis through Receptor-Mediated Events. Mol. Pharmacol. 2008, 74, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Agten, S.M.; Koenen, R.; Ippel, H.; Eckert, V.; von Hundelshausen, P.; Mayo, K.H.; Weber, C.; Hackeng, T.M. Probing Functional Heteromeric Chemokine Protein-Protein Interactions through Conformation-assis ted Oxime-Linkage. Angew. Chem. 2016, 55, 14963–14966. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.L.; Colombo, M.; Gonzales, J.J.; Hollander, W.; Schmid, K. The glycosaminoglycans of the human artery and their changes in atherosclerosis. J. Clin. Investig. 1976, 58, 470. [Google Scholar] [CrossRef] [PubMed]
- Taylor, K.R.; Gallo, R.L. Glycosaminoglycans and their proteoglycans: Host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Handel, T.M.; Johnson, Z.; Crown, S.E.; Lau, E.K.; Proudfoot, A.E. Regulation of protein function by glycosaminoglycans—As exemplified by chemokines. Annu. Rev. Biochem. 2005, 74, 385–410. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sharp, J.S.; Handel, T.M.; Prestegard, J.H. Chemokine oligomerization in cell signaling and migration. Prog. Mol. Biol. Transl. Sci. 2013, 117, 531–578. [Google Scholar] [PubMed]
- Gouwy, M.; Struyf, S.; Berghmans, N.; Vanormelingen, C.; Schols, D.; Van Damme, J. CXCR4 and CCR5 ligands cooperate in monocyte and lymphocyte migration and in inhibition of dual-tropic (R5/X4) HIV-1 infection. Eur. J. Immunol. 2011, 41, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Schiraldi, M.; Struyf, S.; Van Damme, J.; Uguccioni, M. Possible mechanisms involved in chemokine synergy fine tuning the inflammatory response. Immunol. Lett. 2012, 145, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Gouwy, M.; Struyf, S.; Leutenez, L.; Pörtner, N.; Sozzani, S.; Van Damme, J. Chemokines and other GPCR ligands synergize in receptor-mediated migration of monocyte-derived immature and mature dendritic cells. Immunobiology 2014, 219, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Mortier, A.; Van Damme, J.; Proost, P. Overview of the mechanisms regulating chemokine activity and availability. Immunol. Lett. 2012, 145, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Garin, A.; Proudfoot, A.E. Chemokines as targets for therapy. Exp. Cell Res. 2011, 317, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, M.; Cesta, M.C.; Garin, A.; Proudfoot, A.E. Current status of chemokine receptor inhibitors in development. Immunol. Lett. 2012, 145, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Bonvin, P.; Power, C.A. Targeting chemokines: Pathogens can, why can’t we? Cytokine 2015, 74, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.; Power, C.A.; Schwarz, M.K. Anti-chemokine small molecular drugs: A promising future? Expert Opin. Investig. Drugs 2010, 19, 345–355. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Salanga, C.L.; Handel, T.M.; Hamel, D.J. Emerging concepts and approaches for chemokine-receptor drug discovery. Expert Opin. Drug Discov. 2010, 5, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Kufareva, I.; Salanga, C.L.; Handel, T.M. Chemokine and chemokine receptor structure and interactions: Implications for therapeutic strategies. Immunol. Cell Biol. 2015, 93, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Komai, M.; Iwase, M.; Kobayashi, K.; Tahara, H.; Ohshima, E.; Arai, H.; Miki, I. Inihbitory effect of the new orally active CCR4 antagonist K327 on CCR4+CD4+ T cell migration into the lung of mice with ovalbumin-induced lung allergic inflammation. Pharmacology 2009, 84, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Perros, F.; Hoogsteden, H.C.; Coyle, A.J.; Lambrecht, B.N.; Hammad, H. Blockade of CCR4 in a humanized model of asthma reveals a critical role for DC-derived CCL17 and CCL22 in attracting Th2 cells and inducing airway inflammation. Allergy 2009, 64, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Komai, M.; Tanaka, H.; Nagao, K.; Ishizaki, M.; Kajiwara, D.; Miura, T.; Ohashi, H.; Haba, T.; Kawakami, K.; Sawa, E.; et al. A novel CC-chemokine receptor 3 antagonist, Ki19003, inhibits airway eosinophilia and subepithelial/peribronchial fibrosis induced by repeated antigen challenge in mice. J. Pharmacol. Sci. 2010, 112, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Mahler, D.A.; Huang, S.; Tabrizi, M.; Bell, G.M. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: A pilot study. Chest 2004, 126, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Brodmerkel, C.M.; Huber, R.; Covington, M.; Diamond, S.; Hall, L.; Collins, R.; Leffet, L.; Gallagher, K.; Feldman, P.; Collier, P.; et al. Discovery and pharmacological characterization of a novel rodent-active CCR2 antagonist, INCB3344. J. Immunol. 2005, 175, 5370–5378. [Google Scholar] [CrossRef] [PubMed]
- Shahrara, S.; Proudfoot, A.E.; Park, C.C.; Volin, M.V.; Haines, G.K.; Woods, J.M.; Handel, T.M.; Pope, R.M. Inhibition of monocyte chemoattractant protein-1 ameliorates rat adjuvant-induced arthritis. J. Immunol. 2008, 180, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Brühl, H.; Cihak, J.; Schneider, M.A.; Plachý, J.; Rupp, T.; Wenzel, I.; Shakarami, M.; Milz, S.; Ellwart, J.W.; Stangassinger, M.; et al. Dual role of CCR2 during initiation and progression of collagen-induced arthritis: Evidence for regulatory activity of CCR2+ T cells. J. Immunol. 2004, 172, 890–898. [Google Scholar] [CrossRef] [PubMed]
- Struthers, M.; Pasternak, A. CCR2 antagonists. Curr. Top. Med. Chem. 2010, 10, 1278–1298. [Google Scholar] [CrossRef] [PubMed]
- Horuk, R. Chemokine receptor antagonists: Overcoming developmental hurdles. Nat. Rev. Drug Discov. 2008, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; et al. Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.J.; Wang, Y.; Lai, N.; Baumgart, T.; Zhao, B.N.; Dairaghi, D.J.; Bekker, P.; Ertl, L.S.; Penfold, M.E.; Jaen, J.C.; et al. Characterization of CCX282-B, an orally bioavailable antagonist of the CCR9 chemokine receptor, for treatment of inflammatory bowel disease. J. Pharm. Exp. Ther. 2010, 335, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Akashi, S.; Sho, M.; Kashizuka, H.; Hamada, K.; Ikeda, N.; Kuzumoto, Y.; Tsurui, Y.; Nomi, T.; Mizuno, T.; Kanehiro, H.; et al. A Novel Small-Molecule Compound Targeting CCR5 and CXCR3 Prevents Acute and Chronic Allograft Rejection. Transplantation 2005, 80, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Lüttichau, H.R. The herpesvirus 8 encoded chemokines vCCL2 (vMIP-II) and vCCL3 (vMIP-III) target the human but not the murine lymphotactin receptor. Virol. J. 2008, 5, 50. [Google Scholar] [CrossRef] [PubMed]
- Luz, J.G.; Yu, M.; Su, Y.; Wu, Z.; Zhou, Z.; Sun, R.; Wilson, I.A. Crystal Structure of Viral Macrophage Inflammatory Protein I Encoded by Kaposi’s Sarcoma-associated Herpesvirus at 1.7Å. J. Mol. Biol. 2005, 352, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-proteincoupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.S.; Masters, J.; Graham, K.; Liu, L.; Lucas, A.; McFadden, G. Role of the Myxoma Virus Soluble CC-Chemokine Inhibitor Glycoprotein, M-T1, during Myxoma Virus Pathogenesis. Virology 1999, 256, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Bonvin, P.; Dunn, S.M.; Rousseau, F.; Dyer, D.P.; Shaw, J.; Power, C.A.; Handel, T.M.; Proudfoot, A.E. Identification of the pharmacophore of the CC chemokine-binding proteins Evasin-1 and -4 using phage display. J. Biol. Chem. 2014, 289, 31846–31855. [Google Scholar] [CrossRef] [PubMed]
- Takekoshi, T.; Ziarek, J.J.; Volkman, B.F.; Hwang, S.T. A locked, dimeric CXCL12 variant effectively inhibits pulmonary metastasis of CXCR4-expressing melanoma cells due to enhanced serum stability. Mol. Cancer Ther. 2012, 11, 2516–2525. [Google Scholar] [CrossRef] [PubMed]
- Veldkamp, C.T.; Ziarek, J.J.; Peterson, F.C.; Chen, Y.; Volkman, B.F. Targeting SDF-1/CXCL12 with a ligand that prevents activation of CXCR4 through structure-based drug design. J. Am. Chem. Soc. 2010, 132, 7242–7243. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.W.; Liu, Y.; Getschman, A.E.; Peterson, F.C.; Ziarek, J.J.; Li, R.; Volkman, B.F.; Chen, Y. Structural analysis of a novel small molecule ligand bound to the CXCL12 chemokine. J. Med. Chem. 2014, 57, 9693–9699. [Google Scholar] [CrossRef] [PubMed]
- Ziarek, J.J.; Liu, Y.; Smith, E.; Zhang, G.; Peterson, F.C.; Chen, J.; Yu, Y.; Chen, Y.; Volkman, B.F.; Li, R. Fragment-based optimization of small molecule CXCL12 inhibitors for antagonizing the CXCL12/CXCR4 interaction. Curr. Top. Med. Chem. 2012, 12, 2727–2740. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qin, L.; Zacarías, N.V.; de Vries, H.; Han, G.W.; Gustavsson, M.; Dabros, M.; Zhao, C.; Cherney, R.J.; Carter, P.; et al. Structure of CC chemokine receptor 2 with orthosteric and allosteric antagonists. Nature 2016, 540, 458–461. [Google Scholar] [CrossRef] [PubMed]
- Johnson, Z.; Proudfoot, A.E.; Handel, T.M. Interaction of chemokines and glycosaminoglycans: A new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev. 2005, 16, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Nellen, A.; Heinrichs, D.; Berres, M.L.; Sahin, H.; Schmitz, P.; Proudfoot, A.E.; Trautwein, C.; Wasmuth, H.E. Interference with oligomerization and glycosaminoglycan binding of the chemokine CCL5 improves experimental liver injury. PLoS ONE 2012, 7, e36614. [Google Scholar] [CrossRef] [PubMed]
- Ziarek, J.J.; Veldkamp, C.T.; ZhaDyerg, F.; Murray, N.J.; Kartz, G.A.; Liang, X.; Su, J.; Baker, J.E.; Linhardt, R.J.; Volkman, B.F. Heparin oligosaccharides inhibit chemokine (CXC motif) ligand 12 (CXCL12) cardio-protection by binding orthogonal to the dimerization interface, promoting oligomerization, and competing with the chemokine (CXC motif) receptor 4 (CXCR4) N terminus. J. Biol. Chem. 2013, 288, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Thomson, J.M.; Hermant, A.; Jowitt, T.A.; Handel, T.M.; Proudfoot, A.E.; Day, A.J.; Milner, C.M. TSG-6 inhibits neutrophil migration via direct interaction with the chemokine CXCL8. J. Immunol. 2014, 192, 2177–2185. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. https://doi.org/10.3390/ijms18102088
Miller MC, Mayo KH. Chemokines from a Structural Perspective. International Journal of Molecular Sciences. 2017; 18(10):2088. https://doi.org/10.3390/ijms18102088
Chicago/Turabian StyleMiller, Michelle C., and Kevin H. Mayo. 2017. "Chemokines from a Structural Perspective" International Journal of Molecular Sciences 18, no. 10: 2088. https://doi.org/10.3390/ijms18102088
APA StyleMiller, M. C., & Mayo, K. H. (2017). Chemokines from a Structural Perspective. International Journal of Molecular Sciences, 18(10), 2088. https://doi.org/10.3390/ijms18102088