Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents


Abstract
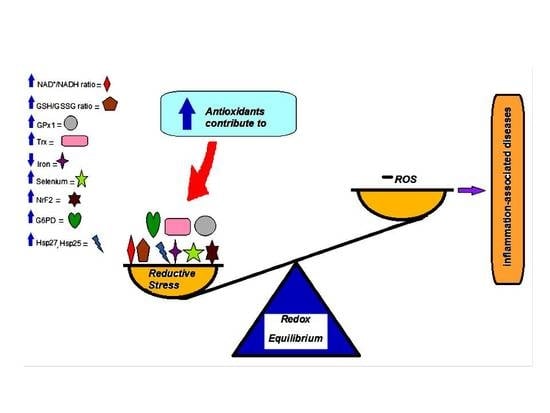
:
1. Introduction
2. Reactive Oxidative Species and Antioxidant Systems
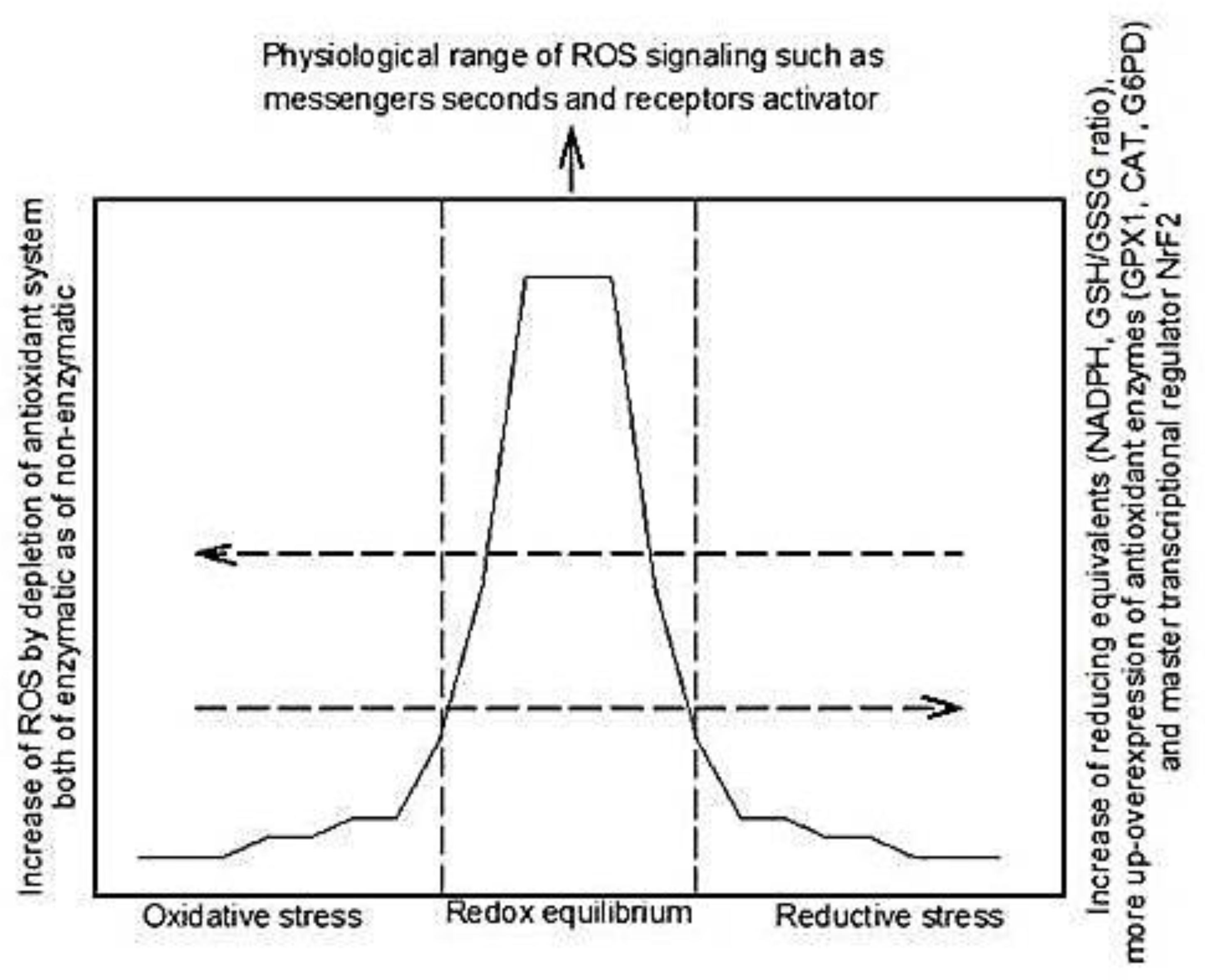
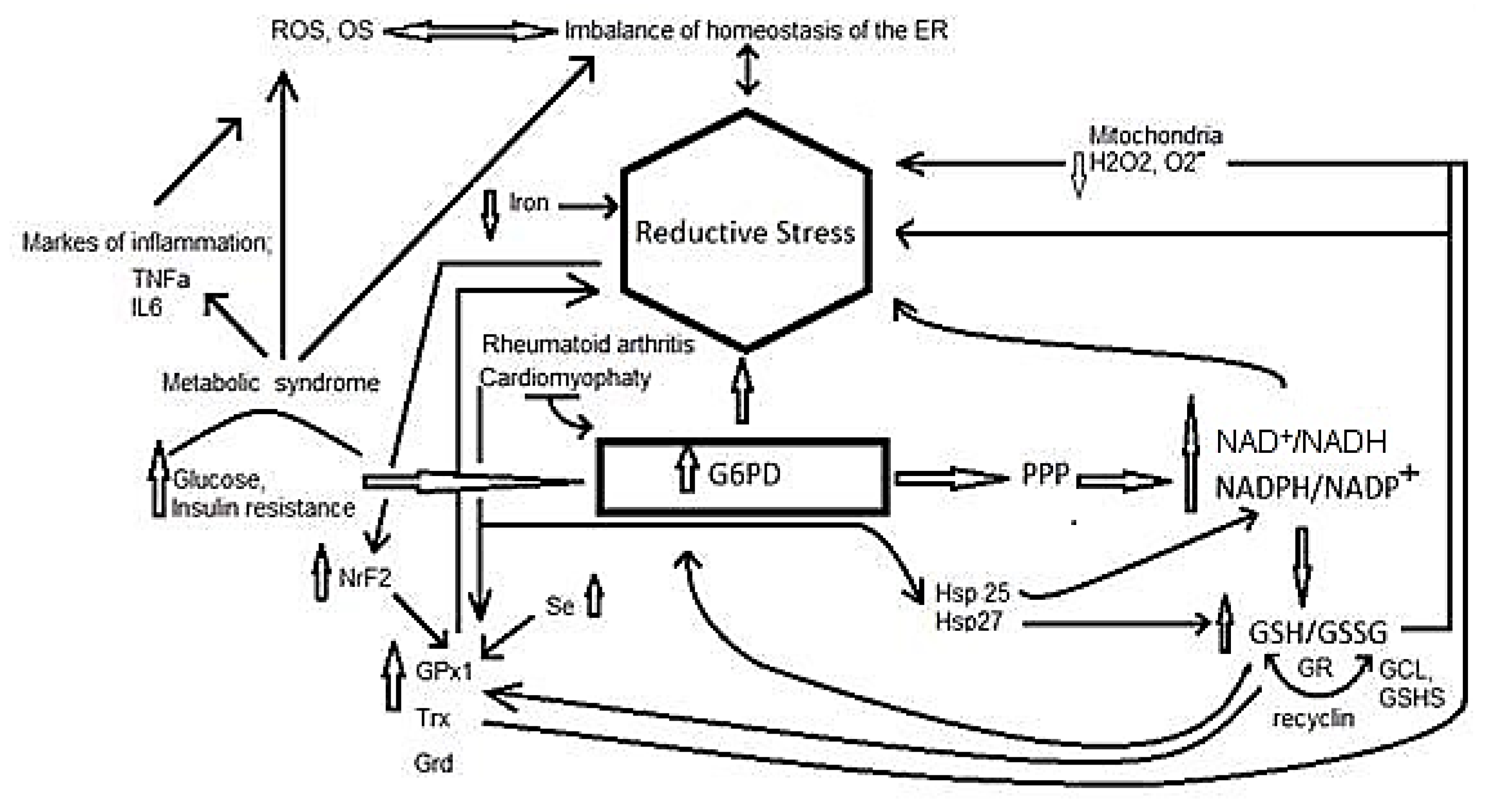
3. Reductive Stress
4. Participation of Different Molecules in Reductive Stress
4.1. Nicotinamide Adenine Dinucleotide oxidized/Nicotinamide Adenine Dinucleotide Reduced Ratio
4.2. Reduced Glutathione/Disulfide Glutathione Ratio
4.3. Glutathione Peroxidase 1 Isoform
4.4. Thiols
4.5. Iron
4.6. Selenium
4.7. Nuclear Erythroid-Related Factor 2
5. Reductive Stress in Inflammation Related Diseases
5.1. Reductive Stress and Cardiac Health
5.2. Reductive Stress and Pulmonary Hypertension
5.3. Reductive Stress and Stent Stenosis
5.4. Reductive Stress and Neuro-Muscular Disorders
5.5. Parkinson’s Disease
5.6. Reductive Stress in Insulin Resistance Associated with Metabolic Syndrome
5.7. Reductive Stress and Rheumatoid Arthritis
5.8. Reductive Stress and Renal Diseases
5.9. Reductive Stress in Infectious Diseases
5.10. Reductive Stress in Alzheimre’s Diseases
6. Situations Inducing Non-Pathological Reductive Stress: Hypoxia and Exercise
7. Adverse Effects of Antioxidant Agents
7.1. Tocopherol
7.2. β-Carotene
7.3. Ascorbic Acid
7.4. N-Acetylcysteine
7.5. Synthetic Antioxidants
7.6. Phenolic Antioxidants
7.7. Estrogens
8. Summary and Conclusions
Author Contributions
Conflicts of Interest
References
- Singh, F.; Charles, A.L.; Schlagowski, A.I.; Bouitbir, J.; Bonifacio, A.; Piquard, F.; Krähenbühl, S.; Geny, B.; Zoll, J. Reductive stress impairs myoblasts mitochondrial function and triggers mitochondrial hormesis. Biochim. Biophys. Acta 2015, 1853, 1574–1585. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Brewer, A.C.; Murray, T.V.; Arno, M.; Zhang, M.; Anilkumar, N.P.; Mann, G.E.; Shah, A.M. Nox4 regulates Nrf2 and glutathione redox in cardiomyocytes in vivo. Free Radic. Biol. Med. 2011, 51, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M.; Stamler, J.S. A central role for S-nitrosylation in apoptosis. Nat. Cell Biol. 2005, 7, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Kang, S.W.; Jeong, W.; Chang, T.S.; Yang, K.S.; Woo, H.A. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr. Opin. Cell Biol. 2005, 17, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.E.; Hare, J.M. Xanthine oxido reductase and cardiovascular disease: Molecular mechanisms and pathophysiological implications. J. Physiol. 2004, 555, 589–606. [Google Scholar] [CrossRef] [PubMed]
- Grune, T. Oxidants and antioxidative defense. Hum. Exp. Toxicol. 2002, 21, 61–62. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Lüpertz, R.; Chovolou, Y.; Kampkötter, A.; Wätjen, W.; Kahl, R. Catalase over expression impairs TNF-α induced NF-κB activation and sensitizes MCF-7 cells against TNF-α. J. Cell. Biochem. 2008, 103, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Guarner-Lans, V.; Zúñiga-Muñoz, A.; Velázquez, E.R.; Cabrera-Orefice, A.; Uribe-Carvajal, S.; Pavón, N. Effect of cross-sex hormonal replacement on antioxidant enzymes in rat retroperitoneal fat adipocytes. Oxid. Med. Cell. Longev. 2016, 2016, 1527873. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative Stress: Oxidants and Antioxidants; Academic Press: London, UK, 1991; ISBN 0-12-642762-3. [Google Scholar]
- Sharapov, M.G.; Goncharov, R.G.; Gordeeva, A.E.; Novoselov, V.I.; Antonova, O.A.; Tikhaze, A.K.; Lankin, V.Z. Enzymatic antioxidant system of endotheliocytes. Dokl. Biochem. Biophys. 2016, 471, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Chirumbolo, S. Role of oxidative stress and antioxidants in daily nutrition and human health. Nutrition 2017, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Christians, E.S.; Benjamin, I.J. Proteostasis and REDOX state in the heart. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H24–H37. [Google Scholar] [CrossRef] [PubMed]
- Trotter, E.W.; Grant, C.M. Thioredoxins are required for protection against a reductive stress in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 2002, 46, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Limphong, P.; Pieper, J.; Liu, Q.; Rodesch, C.K.; Christians, E.; Benjamin, I.J. Glutathione-dependent reductive stress triggers mitochondrial oxidation and cytotoxicity. FASEB J. 2012, 26, 1442–1451. [Google Scholar] [CrossRef] [PubMed]
- Korge, P.; Calmettes, G.; Weiss, J.N. Increased reactive oxygen species production during reductive stress: The roles of mitochondrial glutathione and thioredoxin reductases. Biochim. Biophys. Acta 2015, 1847, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, Y.; Loscalzo, J. Regulation of the protein disulfide proteome by mitochondria in mammalian cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10813–10817. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Rajkumar, A.; Matai, L.; Bhat, A.; Ghosh, A.; Agam, G.; Kaur, S.; Bhatt, N.R.; Mukhopadhyay, A.; Sengupta, S.; et al. Oxidative Homeostasis regulates the response to reductive endoplasmic reticulum stress through translation control. Cell Rep. 2016, 16, 851–865. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.J. Pathogenesis of chronic hyperglycemia: From reductive stress to oxidative stress. J. Diabetes Res. 2014, 2014, 137919. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavinin mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chung, S.S. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [PubMed]
- Lambeth, J.D. NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 2004, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Lee, C.F.; Wang, W.; Karamanlidis, G.; Kuroda, J.; Matsushima, S.; Sadoshima, J.; Tian, R. Elimination of NADPH oxidase activity promotes reductive stress and sensitizes the heart to ischemic injury. J. Am. Heart Assoc. 2014, 3, e000555. [Google Scholar] [CrossRef] [PubMed]
- Rybka, J.; Kupczyk, D.; Kędziora-Kornatowska, K.; Motyl, J.; Czuczejko, J.; Szewczyk-Golec, K.; Kozakiewicz, M.; Pawluk, H.; Carvalho, L.A.; Kędziora, J. Glutathione-related antioxidant defense system in elderly patients treated for hypertension. Cardiovasc. Toxicol. 2011, 11, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Checa, J.C.; Fernández, A.; Morales, A.; Marí, M.; García-Ruiz, C.; Colell, A. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: Lessons from mouse models. CNS Neurol. Disord. Drug Targets 2010, 9, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathionein the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Seelig, G.F.; Simondsen, R.P.; Meister, A. Reversible dissociation of gamma-glutamyl cysteine synthetase in to two subunits. J. Biol. Chem. 1984, 259, 9345–9347. [Google Scholar] [PubMed]
- Hansen, J.M.; Go, Y.M.; Jones, D.P. Nuclear and mitochondrial compartmentation of oxidative stress and redox signaling. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohé, L. Diversity of glutathione peroxidases. Methods Enzymol. 1995, 252, 38–53. [Google Scholar] [PubMed]
- Hu, Y.; Benya, R.V.; Carroll, R.E.; Diamond, A.M. Allelic loss of the gene for the GPX1 selenium-containing protein is a common event in cancer. J. Nutr. 2005, 135, S3021–S3024. [Google Scholar]
- Fisher, A.B. Peroxiredoxin 6: A bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. Antioxid. Redox Signal. 2011, 15, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sies, H.; Lei, X.G. Opposite roles of selenium-dependent glutathione peroxidase-1 insuperoxide generator diquat- and peroxynitrite-induced apoptosis and signaling. J. Biol. Chem. 2001, 276, 43004–43009. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Lubos, E.; Yang, Y.; Galbraith, J.D.; Kelly, N.; Zhang, Y.Y.; Leopold, J.A.; Loscalzo, J. Glutathione peroxidase-1 regulates mitochondrial function to modulate redox-dependent cellular responses. J. Biol. Chem. 2009, 284, 11913–11921. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The Basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Couturier, J.; Przybyla-Toscano, J.; Roret, T.; Didierjean, C.; Rouhier, N. The roles of glutaredoxins ligating Fe-S clusters: Sensing, transfer or repair functions? Biochim. Biophys. Acta 2015, 1853, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Holmgren, A. Thioredoxin and glutaredoxin systems. J. Biol. Chem. 1989, 264, 13963–13966. [Google Scholar] [PubMed]
- Kwon, K.; Kim, J.C. Redox-responsive alginate microsphere containing cystamine. J. Biomater. Sci. Polym. Ed. 2016, 27, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- García-Giménez, J.L.; Romá-Mateo, C.; Pérez-Machado, G.; Peiró-Chova, L.; Pallardó, F.V. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017, 112, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Lodish, H.F.; Kong, N. The secretory pathway is normal in dithiothreitol-treated cells, but disulfide-bonded proteins are reduced and reversibly retained in the endoplasmic reticulum. J. Biol. Chem. 1993, 268, 20598–20605. [Google Scholar] [PubMed]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, S.; Nishimura, A.; Morita, M.; Matsunaga, T.; Abdul-Hamid, H.; Akaike, T. Redox signaling regulated by cysteine persulfide and protein polysulfidation. Molecules 2016, 21, 1721. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.D.; Leibold, E.A. Effects of iron regulatory protein regulation on iron homeostasis during hypoxia. Blood 2003, 102, 3404–3411. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.; Raza, S.T.; Ahmed, F.; Ahmad, A.; Abbas, S.; Mahdi, F. The role of vitamin E in human health and some diseases. Sultan Qaboos Univ. Med. J. 2014, 14, e157–e165. [Google Scholar] [PubMed]
- Reeves, M.A.; Hoffmann, P.R. The human selenoproteome: Recent insights into functions and regulation. Cell. Mol. Life Sci. 2009, 66, 2457–2478. [Google Scholar] [CrossRef] [PubMed]
- Benstoem, C.; Goetzenich, A.; Kraemer, S.; Borosch, S.; Manzanares, W.; Hardy, G.; Stoppe, C. Selenium and its supplementation in cardiovascular disease—What do we know? Nutrients 2015, 7, 3094–3118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Chen, L.; Guo, K.; Zheng, L.; Liu, B.; Yu, W.; Guo, C.; Liu, Z.; Chen, Y.; Tang, Z. Effects of different selenium levels on gene expression of a subset of seleno protein sand antioxidative capacity in mice. Biol. Trace Elem. Res. 2013, 154, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Torres, M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am. J. Respir. Crit. Care Med. 2002, 166, S4–S8. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 improves leptin and insulin resistance provoked by hypothalamic oxidative stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Shelton, L.M.; Park, B.K.; Copple, I.M. Role of Nrf2 in protection against acute kidney injury. Kidney Int. 2013, 84, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Shrestha, O.K.; Ponce, J.M.; Zwerger, M.; Thiemann, D.A.; Young, G.H.; Moore, S.A.; Yu, L.; Lammerding, J.; Wallrath, L.L. Myopathic lamin mutations cause reductive stress and activate the nrf2/keap-1 pathway. PLoS Genet. 2015, 11, e1005231. [Google Scholar] [CrossRef] [PubMed]
- Brewer, A.C.; Mustafi, S.B.; Murray, T.V.; Rajasekaran, N.S.; Benjamin, I.J. Reductive stress linked to small HSPs, G6PD, and Nrf2 pathways in heart disease. Antioxid. Redox Signal. 2013, 18, 1114–14127. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, N.S.; Connell, P.; Christians, E.S.; Yan, L.J.; Taylor, R.P.; Orosz, A.; Zhang, X.Q.; Stevenson, T.J.; Peshock, R.M.; Leopold, J.A.; et al. Human α B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell 2007, 130, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Bauersachs, J.; Widder, J.D. Reductive stress: Linking heat shock protein 27, glutathione, and cardiomyopathy? Hypertension 2010, 55, 1299–1300. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.H.; Min, J.N.; Park, E.M.; Han, M.Y.; Lee, Y.S.; Lee, Y.J.; Park, Y.M. Role of small heat shock protein HSP25 in radio resistance and glutathione-redox cycle. J. Cell. Physiol. 2000, 183, 100–107. [Google Scholar] [CrossRef]
- Rajasekaran, N.S.; Varadharaj, S.; Khanderao, G.D.; Davidson, C.J.; Kannan, S.; Firpo, M.A.; Zweier, J.L.; Benjamin, I.J. Sustained activation of nuclear erythroid 2-related factor 2/antioxidant response element signaling promotes reductive stress in the human mutant protein aggregation cardiomyopathy in mice. Antioxid. Redox Signal. 2011, 14, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Min, X.; Li, C.; Benjamin, I.J.; Qian, B.; Zhang, X.; Ding, Z.; Gao, X.; Yao, Y.; Ma, Y.; et al. Involvement of reductive stress in the cardiomyopathy in transgenic mice with cardiac-specific overexpression of heat shock protein 27. Hypertension 2010, 55, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-mediated increases in l-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B. Limiting reductive stress for treating in-stent stenosis: The heart of the matter? J. Clin. Investig. 2014, 124, 5092–5094. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, M.; Johnson, V.J.; Sharma, R.P. Increase indopamine metabolites inmurines triatum after oral exposure to inorganic but not organic form of selenium. Arch. Environ. Contam. Toxicol. 2000, 39, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Ayaz, M.; Dalkilic, N.; Tuncer, S.; Bariskaner, H. Selenium-induced changes on rats ciatic nerve fibers: Compound action potentials. Methods Find. Exp. Clin. Pharmacol. 2008, 30, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ince, P.G.; Shaw, P.J.; Candy, J.M.; Mantle, D.; Tandon, L.; Ehmann, W.D.; Markesbery, W.R. Iron, selenium and glutathione peroxidase activity are elevated in sporadic motor neuron disease. Neurosci. Lett. 1994, 182, 87–90. [Google Scholar] [CrossRef]
- Greenamyre, J.T.; Cannon, J.R.; Drolet, R.; Mastroberardino, P.G. Lessons from the rotenone model of Parkinson’s disease. Trends Pharmacol. Sci. 2010, 31, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Lloret, A.; Fuchsberger, T.; Giraldo, E.; Vina, J. Reductive stress: A new concept in Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.L.; Siedlak, S.L.; Raina, A.K.; Bautista, J.M.; Smith, M.A.; Perry, G. Increase neuronalglucose-6-phosphate dehydrogenase and sulfhydryl levels indicate reductive compensation to oxidative stress in Alzheimer disease. Arch. Biochem. Biophys. 1999, 370, 236–239. [Google Scholar] [CrossRef] [PubMed]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice over expressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Shen, Y.; Oishi, H.; Matteson, E.L.; Tian, L.; Goronzy, J.J.; Weyand, C.M. Restoring oxidant signaling suppresses proarthritogenic T cell effector functions in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 331ra38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Alexandre, S.; Cao, X.; Lee, A.S. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J. Biol. Chem. 1993, 268, 12003–12009. [Google Scholar] [PubMed]
- Welch, W.J. Mammalian stress response: Cell physiology, structure/function of stress proteins, and implications for medicine and disease. Physiol. Rev. 1992, 72, 1063–1081. [Google Scholar] [PubMed]
- Hilfiker-Kleiner, D.; Landmesser, U.; Drexler, H. Molecular mechanisms in heart failure focus on cardiac hypertrophy, inflammation, angiogenesis, and apoptosis. J. Am. Coll. Cardiol. 2006, 48, A56–A66. [Google Scholar] [CrossRef]
- Chen, Y.; Arrigo, A.P.; Currie, R.W. Heat shock treatment suppresses angiotensin II-induced activation of NF-κB pathway and heart inflammation: A role for IKK depletion by heat shock? Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H1104–H1114. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.B.; Cammarato, A.; Rajasekaran, N.S.; Zhang, H.; Suggs, J.A.; Lin, H.C.; Bernstein, S.I.; Benjamin, I.J.; Golic, K.G. The NADPH metabolic network regulates human αB-crystallin cardiomyopathy and reductive stress in Drosophila melanogaster. PLoS Genet. 2013, 9, e1003544. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, M.; Rajasekaran, N.S. Reductive potential—A savior turns stressor in protein aggregation cardiomyopathy. Biochim. Biophys. Acta 2005, 1852, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P.; Virot, S.; Chaufour, S.; Firdaus, W.; Kretz-Remy, C.; Diaz-Latoud, C. Hsp27 consolidates intracellular redox homeostasis by upholding glutathione in its reduced form and by decreasing iron intracellular levels. Antioxid. Redox Signal. 2005, 7, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zheng, C.; Zhang, Y.; Chang, Y.Z.; Qian, Z.M.; Shen, X. Heat shock protein 27 down regulates the transferrin receptor1-mediated iron uptake. Int. J. Biochem. Cell Biol. 2006, 38, 1402–1416. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Chen, Q.M. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 2017, 327, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Dialynas, G.; Speese, S.; Budnik, V.; Geyer, P.K.; Wallrath, L.L. The role of Drosophila lamin C in muscle function and gene expression. Development 2010, 137, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Ha, K.N.; Chen, Y.; Cai, J.; Sternberg, P. Increased glutathione synthesis through an ARE-Nrf2-dependent pathway by zinc in the RPE: Implication for protection against oxidative stress. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2709–2715. [Google Scholar] [CrossRef] [PubMed]
- Kannan, S.; Muthusamy, V.R.; Whitehead, K.J.; Wang, L.; Gomes, A.V.; Litwin, S.E.; Kensler, T.W.; Abel, E.D.; Hoidal, J.R.; Rajasekaran, N.S. Nrf2 deficiency prevents reductive stress-induced hypertrophic cardiomyopathy. Cardiovasc. Res. 2013, 100, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Yamamoto, M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002, 348, 182–190. [Google Scholar] [PubMed]
- Venardos, K.; Harrison, G.; Headrick, J.; Perkins, A. Effects of dietary selenium on glutathione peroxidase and thioredoxin reductase activity and recovery from cardiac ischemia-reperfusion. J. Trace Elem. Med. Biol. 2004, 18, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P.; Stranges, S.; Griffin, B.A.; Pastor-Barriuso, R.; Guallar, E. Effect of supplementation with high selenium yeast on plasma lipids: A randomized trial. Ann. Intern. Med. 2011, 154, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Laclaustra, M.; Stranges, S.; Navas-Acien, A.; Ordovas, J.M.; Guallar, E. Serum selenium and serum lipids in US adults: National health and nutrition examination Survey (NHANES) 2003–2004. Atherosclerosis 2010, 210, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Gao, S.; Unverzagt, F.W.; Cheng, Y.; Hake, A.M.; Xin, P.; Chen, C.; Liu, J.; Ma, F.; Bian, J.; et al. Selenium level and dyslipidemia in rural elderly Chinese. PLoS ONE 2015, 10, e0136706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta 2006, 1757, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Groth, A.; Vrugt, B.; Brock, M.; Speich, R.; Ulrich, S.; Huber, L.C. Inflammatory cytokines in pulmonary hypertension. Respir. Res. 2004, 15, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, M.; Kamerling, J.P.; Bakker, H.D.; van Gennip, A.H.; Wadman, S.K. l-2-Hydroxyglutaric aciduria: An inborn error of metabolism? J. Inherit. Metab. Dis. 1980, 3, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Sanlioglu, S.; Li, S.; Ritchie, T.; Oberley, L.; Engelhardt, J.F. GPx-1 gene delivery modulates NFκB activation following diverse environmental injuries through a specific subunit of the IKK complex. Antioxid. Redox Signal. 2001, 3, 415–432. [Google Scholar] [CrossRef] [PubMed]
- Papaiahgari, S.; Kleeberger, S.R.; Cho, H.Y.; Kalvakolanu, D.V.; Reddy, S.P. NADPH oxidase and ERK signaling regulates hyperoxia-induced Nrf2-ARE transcriptional response in pulmonary epithelial cells. J. Biol. Chem. 2004, 279, 42302–42312. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, M.; Petucci, C.; Gardell, S.J.; Han, X. Rotenone induces reductive stress and triacylglycerol deposition in C2C12 cells. Int. J. Biochem. Cell Biol. 2013, 45, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- Ammar, E.M.; Couri, D. Acute toxicity of sodium selenite and seleno methionine in mice after ICV or IV administration. Neurotoxicology 1981, 2, 383–386. [Google Scholar] [PubMed]
- Islam, F.; Zia, S.; Sayeed, I.; Kaur, P.; Ahmad, A.S. Effect of selenium on lipids, lipid peroxidation, and sulfhydryl group in neuroendocrine centers of rats. Biol. Trace Elem. Res. 2004, 97, 71–81. [Google Scholar] [CrossRef]
- Zhou, L.Z.; Johnson, A.P.; Rando, T.A. NF κB and AP-1 mediate transcriptional responses to oxidative stress in skeletal muscle cells. Free Radic. Biol. Med. 2001, 31, 1405–1416. [Google Scholar] [CrossRef]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-κB and AP-1 connection: Mechanism of NF-κB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef] [PubMed]
- Teismann, P.; Schulz, J.B. Cellular pathology of Parkinson’s disease: Astrocytes, microglia and inflammation. Cell Tissue Res. 2004, 318, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torres, I.; Roque, P.; El Hafidi, M.; Diaz-Diaz, E.; Baños, G. Association of renal damage and oxidative stress in a rat model of metabolic syndrome. Influence of gender. Free Radic. Res. 2009, 43, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Gerich, J.E.; Meyer, C.; Woerle, H.J.; Stumvoll, M. Renal gluconeo genesis: Its importance in human glucose homeostasis. Diabetes Care 2001, 24, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Hwang, Y.C.; Ananthakrishnan, R.; Oates, P.J.; Guberski, D.; Ramasamy, R. Polyol pathway and modulation of ischemia-reperfusion injury in Type2 diabetic BBZ rat hearts. Cardiovasc. Diabetol. 2008, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Ritov, V.B.; Menshikova, E.V.; Azuma, K.; Wood, R.; Toledo, F.G.; Goodpaster, B.H.; Ruderman, N.B.; Kelley, D.E. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E49–E58. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.L.; Ikeda, Y.; Olsen, G.S.; Busch, A.K.; Mosthaf, L. Insulin signaling is inhibited by micromolar concentrations of H(2)O(2). Evidence for a role of H(2)O(2) in tumor necrosis factor α-mediated insulin resistance. J. Biol. Chem. 1999, 274, 25078–25084. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.D.; Eguchi, S.; Reynolds, C.M.; Eguchi, K.; Frank, G.D.; Motley, E.D. Hydrogen peroxide inhibits insulin signaling in vascular smooth muscle cells. Exp. Biol. Med. 2003, 228, b836–b842. [Google Scholar] [CrossRef]
- Mahadev, K.; Zilbering, A.; Zhu, L.; Goldstein, B.J. Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade. J. Biol. Chem. 2001, 276, 21938–21942. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.R.; Sampaio, I.H.; Teodoro, B.G.; Sousa, T.A.; Zoppi, C.C.; Queiroz, A.L.; Passos, M.A.; Alberici, L.C.; Teixeira, F.R.; Manfiolli, A.O.; et al. Hydrogen peroxide production regulates the mitochondrial function in insulin resistant muscle cells: Effect of catalase overexpression. Biochim. Biophys. Acta 2013, 1832, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Sousa, V.C.; Carmo, R.F.; Vasconcelos, L.R.; Aroucha, D.C.; Pereira, L.M.; Moura, P.; Cavalcanti, M.S. Association of catalase and glutathione peroxidase1 polymorphisms with chronic hepatitis c outcome. Ann. Hum. Genet. 2016, 80, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Faucher, K.; Rabinovitch-Chable, H.; Cook-Moreau, J.; Barrière, G.; Sturtz, F.; Rigaud, M. Overexpression of human GPX1 modifies Bax to Bcl-2 apoptotic ratio in human endothelial cells. Mol. Cell. Biochem. 2005, 277, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.R.; Barrett, J.C. Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 2002, 21, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Mirochnitchenko, O.; Palnitkar, U.; Philbert, M.; Inouye, M. Thermosensitive phenotype of transgenic mice overproducing human glutathione peroxidases. Proc. Natl. Acad. Sci. USA 1995, 92, 8120–8124. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Real, J.M.; Ricart, W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr. Rev. 2003, 24, 278–301. [Google Scholar] [CrossRef] [PubMed]
- Mathews, S.T.; Kim, T. A novel regulator of insulin action: Role in insulin resistance, chapter 3. In Insulin Resistance: New Research; Yao, E.B., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2009; pp. 99–116. [Google Scholar]
- Schwartz, E.A.; Reaven, P.D. Molecular and signaling mechanisms of atherosclerosis in insulin resistance. Endocrinol. Metab. Clin. N. Am. 2006, 35, 525–549. [Google Scholar] [CrossRef] [PubMed]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Jager, J.; Grémeaux, T.; Cormont, M.; Le Marchand-Brustel, Y.; Tanti, J.F. Interleukin-1-beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 2007, 148, 241–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. MetabolicregulationofT lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Halleck, M.M.; Holbrook, N.J.; Skinner, J.; Liu, H.; Stevens, J.L. The molecular response o reductive stress in LLC-PK1 renal epithelial cells: Coordinate transcriptional regulation of gadd153 and grp78 genes by thiols. Cell Stress Chaperones 1997, 2, 31–40. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lee, A.S. Transcriptional activation of the glucose-regulated protein genes and their heterologous fusion genes by beta-mercaptoethanol. Mol. Cell. Biol. 1987, 7, 2974–2976. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Mavi, P.S.; Bhatt, D.; Kumar, A. Thiol reductive stress induces cellulose-anchored biofilm formation in Mycobacterium tuberculosis. Nat. Commun. 2016, 7, 11392. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.L. Envelope stress responses and Gram-negative bacterial pathogenesis. Mol. Microbiol. 2005, 56, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Badía, M.C.; Giraldo, E.; Dasí, F.; Alonso, D.; Lainez, J.M.; Lloret, A.; Viña, J. Reductive stress in young healthy individuals at risk of Alzheimer disease. Free Radic. Biol. Med. 2013, 63, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Margaritelis, N.V.; Kyparos, A.; Paschalis, V.; Theodorou, A.A.; Panayiotou, G.; Zafeiridis, A.; Dipla, K.; Nikolaidis, M.G.; Vrabas, I.S. Reductive stress after exercise: The issue of redox individuality. Redox Biol. 2014, 2, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, Z.; Lappalainen, J.; Oksala, N.K.; Laaksonen, D.E.; Khanna, S.; Sen, C.K.; Atalay, M. Diabetes impairs exercise training-associated thioredoxin response and glutathione status in rat brain. J. Appl. Physiol. 1985, 106, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Lappalainen, J.; Laaksonen, D.E.; Oksala, K.J.; Khanna, S.; Sen, C.K.; Atalay, M. Acute exercise and thioredoxin-1 in rat brain, and α-lipoic acid and thioredoxin-interacting protein response, in diabetes. Int. J. Sport Nutr. Exerc. Metab. 2010, 20, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Gudkov, A.V.; Komarova, E.A. p53 and the carcinogenicity of chronic inflammation. Cold Spring Harb. Perspect. Med. 2016, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Joyce, A.; Ingold, K.U. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes? Arch. Biochem. Biophys. 1983, 221, 281–290. [Google Scholar] [CrossRef]
- Burton, G.W.; Joyce, A.; Ingold, K.U. First proof that vitamin E is major lipid-soluble, chain-breaking antioxidant in human blood plasma. Lancet 1982, 2, 327. [Google Scholar] [CrossRef]
- Smith, B.J.; Lucas, E.A.; Turner, R.T.; Evans, G.L.; Lerner, M.R.; Brackett, D.J.; Stoecker, B.J.; Arjmandi, B.H. Vitamin E provides protection for bone in mature hind limb unloaded male rats. Calcif. Tissue Int. 2005, 76, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Iwaniec, U.T.; Turner, R.T.; Smith, B.J.; Stoecker, B.J.; Rust, A.; Zhang, B.; Vasu, V.T.; Gohil, K.; Cross, C.E.; Traber, M.G. Evaluation of long-term vitamin E insuffciency or excess on bone mass, density, and microarchitecture in rodents. Free Radic. Biol. Med. 2013, 65, 1209–1214. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-analysis: High-dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med. 2005, 142, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Terao, J.; Matsushita, S. The peroxidizing effect of α-tocopherol on autoxidation of methyl linoleate in bulk phase. Lipids 1986, 21, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.J.; Xiang, Y.B.; Yang, G.; Li, H.L.; Lan, Q.; Gao, Y.T.; Zheng, W.; Shu, X.O.; Fowke, J.H. Vitamin E intake and the lung cancer risk among female nonsmokers: A report from the shanghai women’s health study. Int. J. Cancer 2015, 136, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Slatore, C.G.; Littman, A.J.; Au, D.H.; Satia, J.A.; White, E. Long-term use of supplemental multivitamins, vitamin C, vitamin E, and folate does not reduce the risk of lung cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 524–530. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, A.R.; Larrick, J.W. Paradoxical effects of antioxidants on cancer. Rejuvenation Res. 2014, 17, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra15. [Google Scholar] [CrossRef] [PubMed]
- Goodman, G.E.; Thornquist, M.; Kestin, M.; Metch, B.; Anderson, G.; Omenn, G.S. The association between participant characteristics and serum concentrations of beta-carotene, retinol, retinyl palmitate, and a α-tocopherol among participants in the carotene and retinol efficacy trial (CARET) for prevention of lung cancer. Cancer Epidemiol. Biomark. Prev. 1996, 5, 815–821. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Sao Paulo Med. J. 2015, 133, 164–165. [Google Scholar] [CrossRef] [PubMed]
- MacLennan, R.; Macrae, F.; Bain, C.; Battistutta, D.; Chapuis, P.; Gratten, H.; Lambert, J.; Newland, R.C.; Ngu, M.; Russell, A.; et al. Randomized trial of intake of fat, fiber, and beta carotene to prevent colorectal adenomas. J. Natl. Cancer Inst. 1995, 87, 1760–1766. [Google Scholar] [CrossRef] [PubMed]
- Hatch, M.; Mulgrew, S.; Bourke, E.; Keogh, B.; Costello, J. Effect of mega doses of ascorbic acid on serum and urinary oxalate. Eur. Urol. 1980, 6, 166–169. [Google Scholar] [PubMed]
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 392, 559. [Google Scholar] [CrossRef] [PubMed]
- Branen, A.L. Toxicology and biochemistry of butylated hydroxyanisole and butylated hydroxytoluene. J. Am. Oil Chem. Soc. 1975, 52, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Fukushima, S.; Hagiwara, A.; Shibata, M.; Ogiso, T. Carcinogenicity of butylated hydroxyanisole in F344 rats. J. Natl. Cancer Inst. 1983, 70, 343–352. [Google Scholar] [PubMed]
- Thompson, D.C.; Cha, Y.N.; Trush, M.A. The peroxidase-dependent activation of butylated hydroxyanisole and butylated hydroxytoluene (BHT) to reactive intermediates. Formation of BHT-quinone methide via a chemical-chemical interaction. J. Biol. Chem. 1989, 264, 3957–3965. [Google Scholar] [PubMed]
- Li, X.; Cao, S.; Mao, B.; Bai, Y.; Chen, X.; Wang, X.; Wu, Y.; Li, L.; Lin, H.; Lian, Q.; et al. Effects of butylated hydroxyanisole on the steroidogenesis of rat immature Leydig cells. Toxicol. Mech. Methods 2016, 26, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Hodnick, W.F.; Kung, F.S.; Roettger, W.J.; Bohmont, C.W.; Pardini, R.S. Inhibition of mitochondrial respiration and production of toxic oxygen radicals by flavonoids. A structure-activity study. Biochem. Pharmacol. 1986, 35, 2345–2357. [Google Scholar] [CrossRef]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A. Resveratrol alters human endothelial cells redox state and causes mitochondrial-dependent cell death. Food Chem. Toxicol. 2015, 78, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Posadino, A.M.; Cossu, A.; Giordo, R.; Zinellu, A.; Sotgia, S.; Vardeu, A.; Hoa, P.T.; Deiana, L.; Carru, C.; Pintus, G. Coumaricacid induces mitochondrialdamage and oxidative-mediatedcell death of human endothelial cells. Cardiovasc. Toxicol. 2013, 13, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Ayres, S.; Abplanalp, W.; Liu, J.H.; Subbiah, M.T. Mechanisms involved in the protective effect of estradiol-17beta on lipid peroxidation and DNA damage. Am. J. Physiol. 1998, 274, E1002–E1008. [Google Scholar] [PubMed]
- Bednarek-Tupikowska, G. Antioxidant properties of estrogens. Ginekol. Pol. 2002, 73, 61–67. [Google Scholar] [PubMed]
- Lee, Y.; Kim, M.; Choi, K.; Kim, J.; Bae, W.; Kim, S.; Sohn, C. Relationship between inflammation biomarkers, antioxidant vitamins, and bone mineral density in patients with metabolic syndrome. Nutr. Res. Pract. 2011, 5, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Akçakaya, H.; Tok, S.; Dal, F.; Cinar, S.A.; Nurten, R. β-carotene treatment alters the cellular death process in oxidative stress-induced K562 cells. Cell Biol. Int. 2017, 41, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, V.O.; Grattagliano, I.; Portincasa, P.; Palasciano, G. Systemic oxidative alterations are associated with visceral adiposity and liver steatosis in patients with metabolic syndrome. J. Nutr. 2006, 136, 3022–3026. [Google Scholar] [PubMed]
- Yeh, S.L.; Wang, H.M.; Chen, P.Y.; Wu, T.C. Interactions of beta-carotene and flavonoids on the secretion of pro-inflammatory mediators in an in vitro system. Chem. Biol. Interact. 2009, 179, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Folsom, A.R.; Harnack, L.; Halliwell, B.; Jacobs, D.R. Does supplemental vitamin C increase cardiovascular disease risk in women with diabetes? Am. J. Clin. Nutr. 2004, 80, 1194–1200. [Google Scholar] [PubMed]
- Mohammed, B.M.; Sanford, K.W.; Fisher, B.J.; Martin, E.J.; Contaifer, D.; Warncke, U.O.; Wijesinghe, D.S.; Chalfant, C.E.; Brophy, D.F.; Fowler, A.A.; et al. Impact of high dose vitamin C on platelet function. World J. Crit. Care Med. 2017, 6, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Hamishehkar, H.; Ranjdoost, F.; Asgharian, P.; Mahmoodpoor, A.; Sanaie, S. Vitamins, are they safe? Adv. Pharm. Bull. 2016, 6, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Mashour, S.; Turner, J.F.; Merrell, R. Acute renal failure, oxalosis, and vitamin C supplementation: A case report and review of the literature. Chest 2000, 118, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Yaich, S.; Chaabouni, Y.; Charfeddine, K.; Zaghdane, S.; Kharrat, M.; Kammoun, K.; Makni, S.; Boudawara, T.; Hachicha, J. Secondary oxalosis due to excess vitamin C intake: A cause of graft lossin a renal transplant recipient. Saudi J. Kidney Dis. Transplant. 2014, 25, 113–116. [Google Scholar] [CrossRef]
- Wahlqvist, M.L. Antioxidant relevance to human health. Asia Pac. J. Clin. Nutr. 2013, 22, 171–176. [Google Scholar] [PubMed]
- Baysal, E.; Sullivan, S.G.; Stern, A. Prooxidant and antioxidant effects of ascorbate on tBuOOH-induced erythrocyte membrane damage. Int. J. Biochem. 1989, 21, 1109–1113. [Google Scholar] [CrossRef]
- Bachowski, G.J.; Thomas, J.P.; Girotti, A.W. Ascorbate-enhanced lipid peroxidation in photooxidized cell membranes: Cholesterol product analysis as a probe of reaction mechanism. Lipids 1988, 23, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Kondakçı, G.; Aydın, A.F.; Doğru-Abbasoğlu, S.; Uysal, M. The effect of N-acetylcysteine supplementation on serum homocysteine levels and hepatic and renal oxidative stress in homocysteine thiolactone-treated rats. Arch. Physiol. Biochem. 2017, 123, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Witschi, H.; Malkinson, A.M.; Thompson, J.A. Metabolism and pulmonary toxicity of butylated hydroxytoluene (BHT). Pharmacol. Ther. 1989, 42, 89–113. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tayama, K.; Nakao, T.; Hiraga, K. On the mechanism of butylated hydroxytoluene-induced hepatic toxicity in rats. Biochem. Pharmacol. 1984, 33, 2669–2674. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Tayama, K. Nephrotoxicity of butylated hydroxytoluene in phenobarbital-pretreated male rats. Arch. Toxicol. 1988, 61, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Sevestre, H.; Ibe, B.O.; Thompson, J.A. Formation and reactivity of alternative quinone methides from butylated hydroxytoluene: Possible explanation for species-specific pneumotoxicity. Chem. Res. Toxicol. 1990, 3, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Hirose, M.; Masuda, A.; Hasegawa, R.; Wada, S.; Ito, N. Regression of butylated hydroxyanisole (BHA)-induced hyperplasia but not dysplasia in the forestomach of hamsters. Carcinogenesis 1990, 11, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M. Epigenetic promoting effects of butylated hydroxyanisole. Food Chem. Toxicol. 1986, 24, 1163–1166. [Google Scholar] [CrossRef]
- Wätjen, W.; Michels, G.; Steffan, B.; Niering, P.; Chovolou, Y.; Kampkötter, A. Low concentrations of flavonoids are protective in rat H4IIE cells whereas high concentrations cause DNA damage and apoptosis. J. Nutr. 2005, 135, 525–531. [Google Scholar] [PubMed]
- Sergediene, E.; Jönsson, K.; Szymusiak, H.; Tyrakowska, B.; Rietjens, I.M.; Cenas, N. Prooxidant toxicity of polyphenolic antioxidants to HL-60 cells: Description of quantitative structure-activity relationships. FEBS Lett. 1999, 462, 392–396. [Google Scholar] [CrossRef]
- Robaszkiewicz, A.; Balcerczyk, A.; Bartosz, G. Antioxidative and prooxidative effects of quercetin on A549 cells. Cell Biol. Int. 2007, 31, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Decker, E.A. Phenolics: Prooxidants or antioxidants? Nutr. Rev. 1997, 55, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Passi, S.; Picardo, M.; Nazzaro-Porro, M. Comparative cytotoxicity of phenols in vitro. Biochem. J. 1987, 245, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Laughton, M.J.; Halliwell, B.; Evans, P.J.; Hoult, J.R. Antioxidant and pro-oxidant actions of the plant phenolics quercetin, gossypol and myricetin: Effects on lipid peroxidation, hydroxyl radical generation and bleomycin-dependent damage to DNA. Biochem. Pharmacol. 1989, 38, 2859–2865. [Google Scholar] [CrossRef]
- Sousa, R.L.; Marletta, M.A. Inhibition of cytochrome P-450 activity in rat liver microsomes by the naturally occurring flavonoid, quercetin. Arch. Biochem. Biophys. 1985, 240, 345–357. [Google Scholar] [CrossRef]
- Zúñiga-Muñoz, A.M.; Guarner, V.; Díaz-Cruz, A.; Diaz-Diaz, E.; Nava-Cuellar, C.; Beltrán-Rodríguez, U.; Peres-Torres, I. Modulation of oxidative stress in fatty Liver of rat with metabolic syndrome by hibiscus sabdariffa. Immunol. Endocr. Metab. Agents Med. Chem. 2013, 3, 196–205. [Google Scholar]
- Castrejón-Tellez, V.; Rodríguez-Pérez, J.M.; Pérez-Torres, I.; Pérez-Hernández, N.; Cruz-Lagunas, A.; Guarner-Lans, V.; Vargas-Alarcón, G.; Rubio-Ruiz, M.E. the effect of resveratrol and quercetin treatment on ppar mediated uncoupling protein (ucp-) 1, 2, and 3 expression in visceral white adipose tissue from metabolic syndrome rats. Int. J. Mol. Sci. 2016, 17, 1069. [Google Scholar] [CrossRef] [PubMed]
- Pasciu, V.; Posadino, A.M.; Cossu, A.; Sanna, B.; Tadolini, B.; Gaspa, L.; Marchisio, A.; Dessole, S.; Capobianco, G.; Pintus, G. Akt downregulation by flavin oxidase-induced ROS generation mediates dose-dependent endothelial cell damage elicited by natural antioxidants. Toxicol. Sci. 2010, 114, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Giordo, R.; Cossu, A.; Pasciu, V.; Hoa, P.T.; Posadino, A.M.; Pintus, G. Different redox response elicited by naturally occurring antioxidants in human endothelial cells. Open Biochem. J. 2013, 7, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.C.; Zuñiga-Muñoz, A.; Guarner Lans, V.; Díaz-Díaz, E.; Tena Betancourt, C.A.; Pérez-Torres, I. Modulation of the activities of catalase, cu-zn, mn superoxide dismutase, and glutathione peroxidase in adipocyte from ovariectomised female rats with metabolic syndrome. Int. J. Endocrinol. 2014, 2014, 175080. [Google Scholar] [CrossRef] [PubMed]
- Liehr, J.G.; Roy, D. Free radical generation by redox cycling of estrogens. Free Radic. Biol. Med. 1990, 8, 415–423. [Google Scholar] [CrossRef]
- Rivera-Portalatin, N.M.; Vera-Serrano, J.L.; Prokai-Tatrai, K.; Prokai, L. Comparison of estrogen-derived ortho-quinone and para-quinol concerning induction of oxidative stress. J. Steroid Biochem. Mol. Biol. 2007, 105, 71–75. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Disease | Source of RS | References |
|---|---|---|
| Cardiomyopathy | ↑ GSH/GSSG ratio | Rajasekaran et al., 2007 [60]; Bauersachs et al., 2010 [61]; Brewer et al., 2013 [59]; Baek et al., 2000 [62]; Rajasekaran et al., 2011 [63] |
| ↓ Free iron content | Zhang et al., 2010 [64] | |
| Pulmonary hypertension | ↑ NADPH/NADP+ ratio | Oldham et al., 2015 [65] |
| Stent stenosis | ↑ GSH/GSSG ratio ↑ NADPH/NADP+ ratio | de Haan., 2014 [66] |
| Muscular dystrophy | ↑ GSH/GSSG ratio | Rajasekaran et al., 2007 [60]; Dialynas et al., 2015 [58] |
| Neurological disorders | ↑ Selenium levels | Tsunoda et al., 2000 [67]; Ayaz et al., 2008 [68] |
| ↑ GPx activity | Ince et al., 1994 [69] | |
| Parkinson’s disease | ↑ NADH/NAD+ ratio | Greenamyre et al., 2010 [70] |
| Alzheimer’s disease | ↑ G6PD and GSH | Lloret et al., 2016 [71]; Russell et al., 1999 [72] |
| Metabolic syndrome and insulin resistance | ↑ GPx1 expression ↑ NADPH/NADP+ ratio | McClung et al., 2004 [73] |
| Rheumatoid arthritis | ↑ NADPH/NADP+ ratio | Yang et al., 2016 [74] |
| Renal diseases | ↑ GSH/GSSG ratio | Li et al., 1993 [75] |
| ↑ Thiols | Welch et al., 1992 [76] | |
| Cancer | ↑ NADH/NAD+ ratio | Oldham et al., 2015 [66] |
| Antioxidant Agent | Mechanisms | Associated Pathology | References |
|---|---|---|---|
| Tocopherol or Vitamin E | Pro-oxidant activity by Fenton reaction | Bone alterations lung cancer | Smith et al., 2005 [132]; Iwaniec et al., 2013 [133]; Wu et al., 2015 [136]; Slatore et al., 2008 [137] |
| NAC | Reduction of NAD+/NADH ratio | Cardiovascular disorders Lung cancer | Zhang et al., 2012 [17]; Mendelsohn et al., 2014 [138]; Sayin et al., 2014 [139] |
| β-carotene | Pro-oxidant and pro-inflammatory | Cancer Colorectal polyps | Goodman et al., 1996 [140]; Bjelakovic et al., 2015 [141] MacLennan et al., 1995 [142] |
| Ascorbic acid (Vitamin C) | Pro-oxidant activity by Fenton reaction | Renal calcium oxalate deposition DNA damage of lymphocytes | Hatch et al., 1980 [143]; Podmore et al., 1998 [144] |
| BHA and BHT | Pro-oxidative properties | Cancer Pulmonary toxicity Reproductive damage | Branen, 1975 [145]; Ito et al., 1983 [146]; Thompson et al., 1989 [147] Li et al., 2016 [148] |
| Flavonoids | Pro-oxidant activity by Fenton reaction | DNA damage, Apoptosis Hypertension | Hodnick et al., 1986 [149] |
| Resveratrol | Pro-oxidant by CYP2C9 | Endothelial cell death | Posadino et al., 2015 [150] |
| Coumaric Acid | Pro-oxidant Mitochondrial Damage | Endothelial cell death | Posadino et al., 2013 [151] |
| Estrogens | Pro-oxidative properties | Cell damage Breast cancer | Ayres et al., 1998 [152] Bednarek, 2002 [153] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Torres, I.; Guarner-Lans, V.; Rubio-Ruiz, M.E. Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. Int. J. Mol. Sci. 2017, 18, 2098. https://doi.org/10.3390/ijms18102098
Pérez-Torres I, Guarner-Lans V, Rubio-Ruiz ME. Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. International Journal of Molecular Sciences. 2017; 18(10):2098. https://doi.org/10.3390/ijms18102098
Chicago/Turabian StylePérez-Torres, Israel, Verónica Guarner-Lans, and María Esther Rubio-Ruiz. 2017. "Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents" International Journal of Molecular Sciences 18, no. 10: 2098. https://doi.org/10.3390/ijms18102098
APA StylePérez-Torres, I., Guarner-Lans, V., & Rubio-Ruiz, M. E. (2017). Reductive Stress in Inflammation-Associated Diseases and the Pro-Oxidant Effect of Antioxidant Agents. International Journal of Molecular Sciences, 18(10), 2098. https://doi.org/10.3390/ijms18102098