DNA Adducts Formed by Aristolochic Acid Are Unique Biomarkers of Exposure and Explain the Initiation Phase of Upper Urothelial Cancer


Abstract
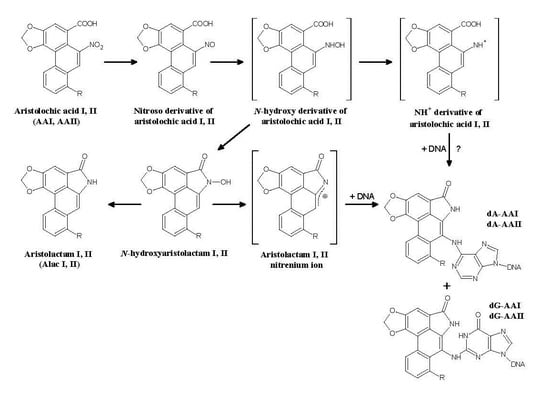
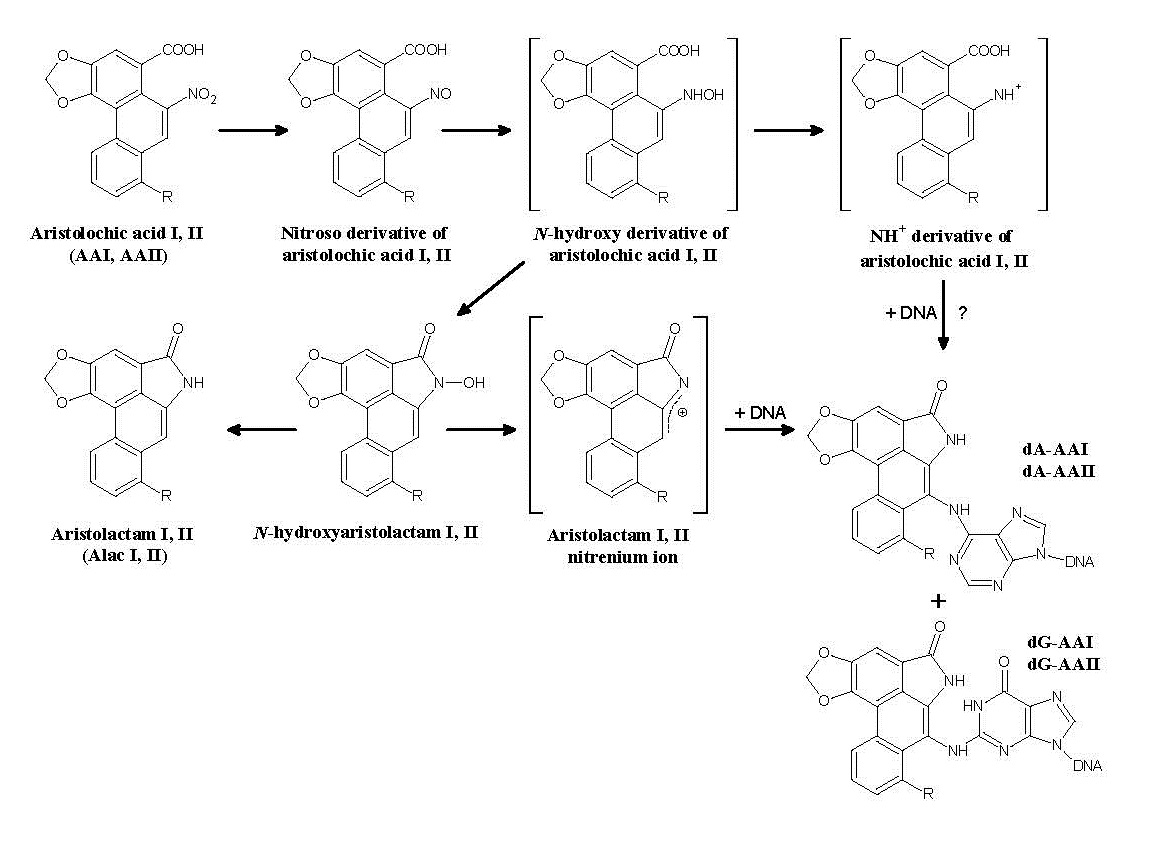
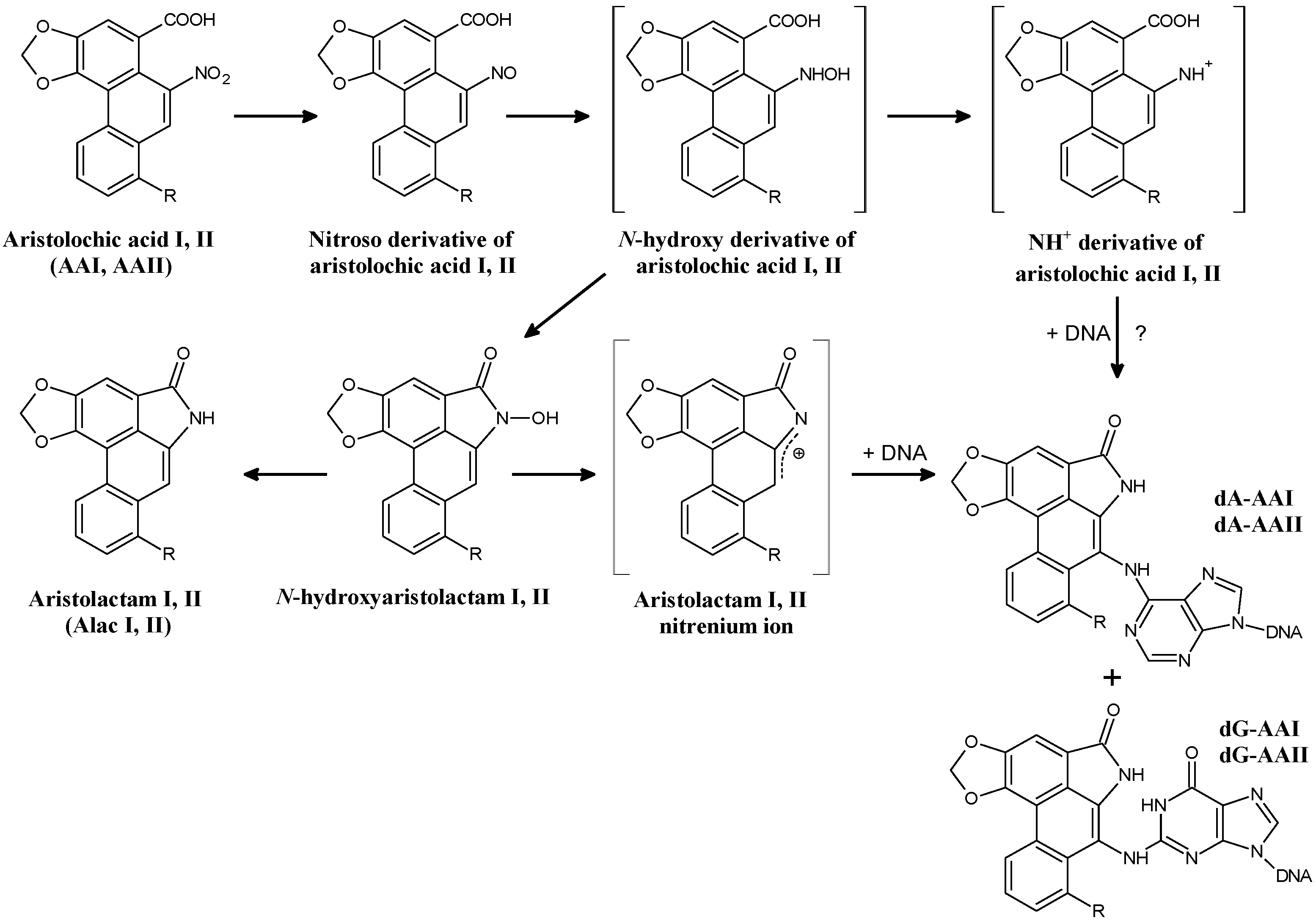
:
1. Introduction
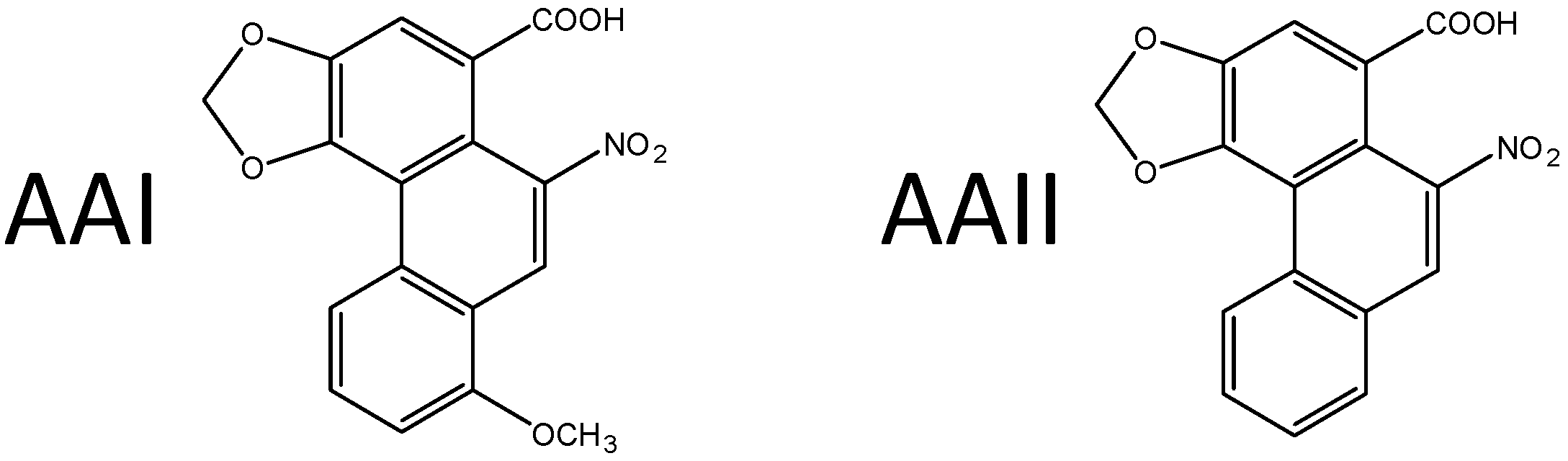
2. Aristolochic Acid
2.1. Aristolochic Acid (AA) as a Carcinogen Causing Upper Urothelial Cancer in Aristolochic Acid Nephropathy (AAN) and Balkan Endemic Nephropathy (BEN) Patients and Renal Cell Carcinoma in Certain Other Human Populations
2.1.1. AA-Derived DNA Adducts and Their Role in the Initiation of Upper Urothelial Cancer
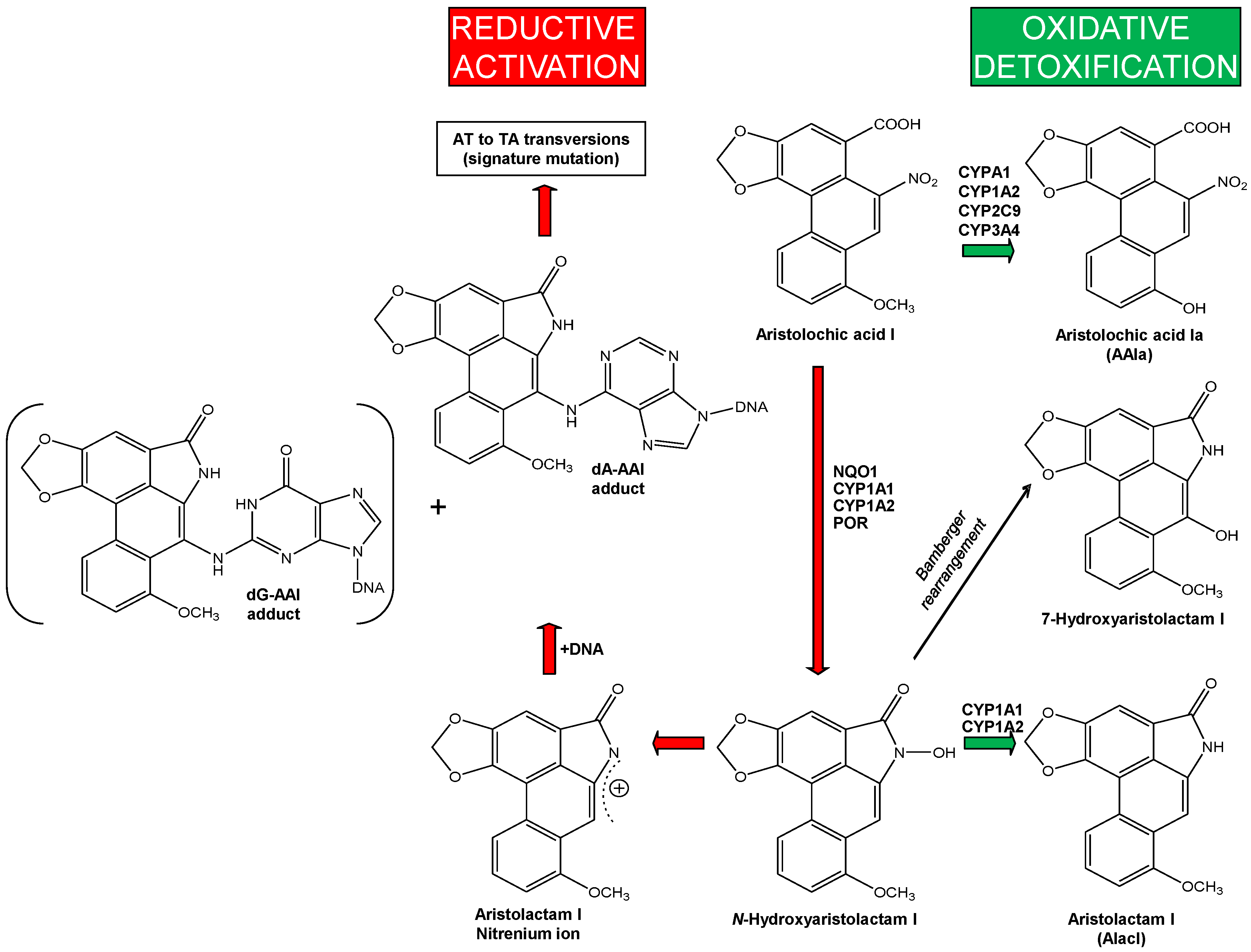
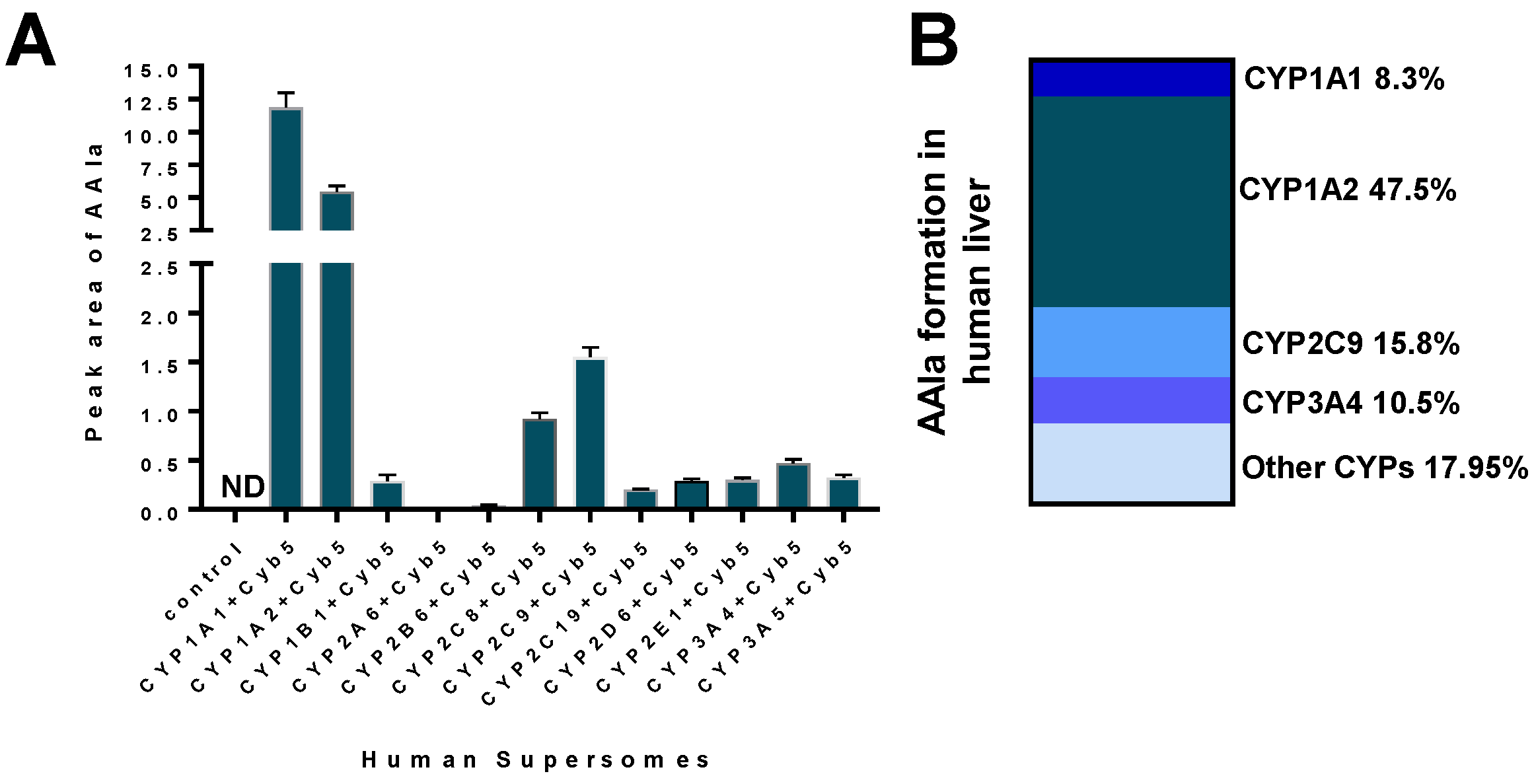
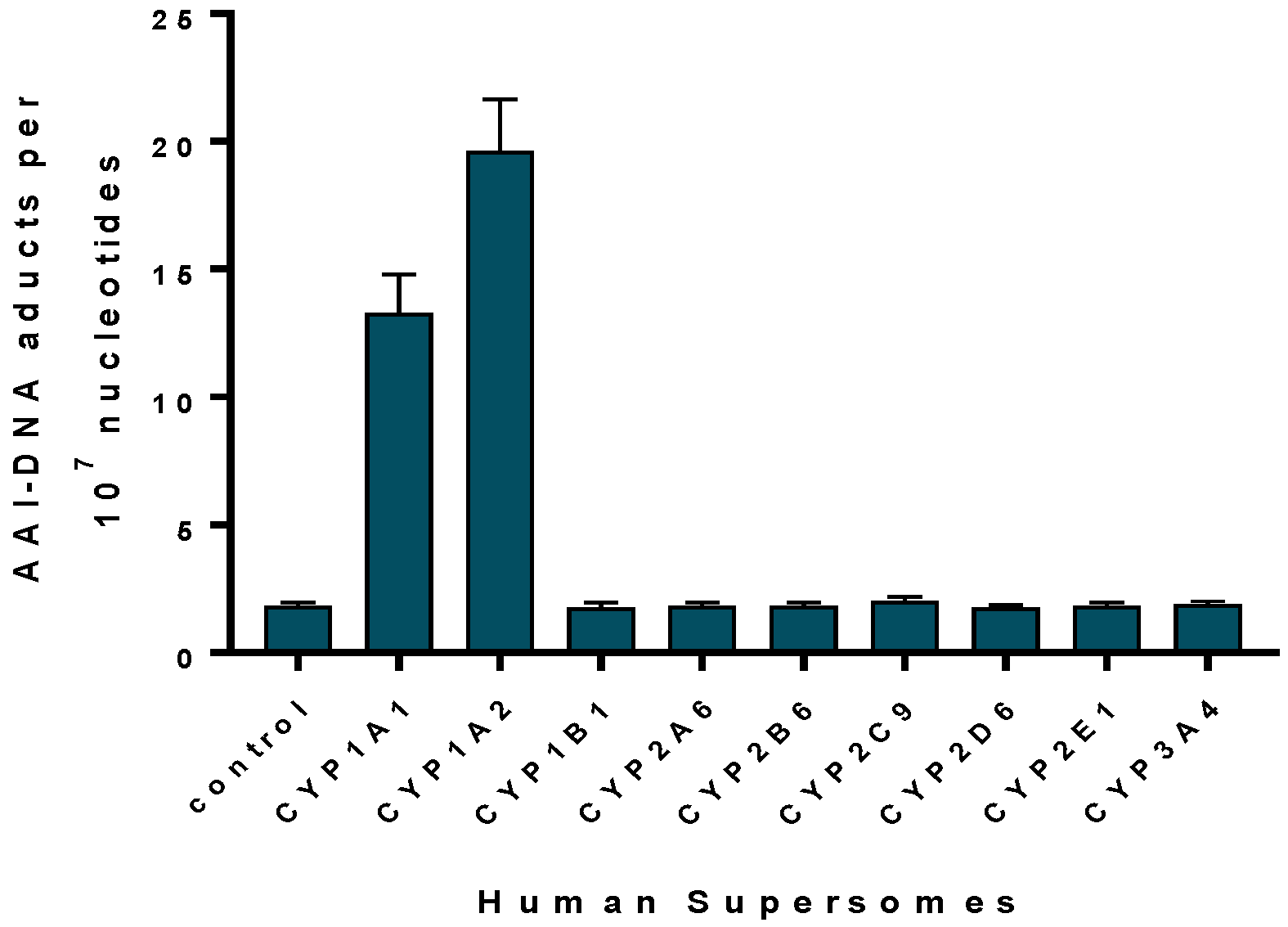
2.1.2. Mechanisms of Enzymatic Activation of AA to Metabolites Forming AA-Derived DNA Adducts and Its Detoxification Resulting in Attenuation of AA-Mediated Diseases
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poirier, M. Chemical-induced DNA damage and human cancer risk. Nat. Rev. Cancer 2004, 4, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.C. Chemical-induced DNA damage and human cancer risk. Discov. Med. 2012, 14, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Poirier, M.C. Linking DNA adduct formation and human cancer risk in chemical carcinogenesis. Environ. Mol. Mutagen. 2016, 57, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Harris, C.C. Advances in chemical carcinogenesis: A historical review and prospective. Cancer Res. 2008, 68, 6863–6872. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Metabolism of chemical carcinogens. Carcinogenesis 2000, 21, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H. DNA adducts as markers of exposure and risk. Mutat. Res. 2005, 577, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H. The Formation of DNA Adducts. In Cancer Handbook; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar]
- Phillips, D.H.; Arlt, V.M. Genotoxicity: Damage to DNA and its consequences. EXS 2009, 99, 87–110. [Google Scholar] [PubMed]
- Baird, W.M.; Hooven, L.A.; Mahadevan, B. Carcinogenic polycyclic aromatic hydrocarbon-DNA adducts and mechanism of action. Environ. Mol. Mutagen. 2005, 45, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J.; le Marchand, L. Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: Lessons learned from aromatic amines. Chem. Res. Toxicol. 2011, 24, 1169–1214. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Arlt, V.M. The 32P-postlabeling assay for DNA adducts. Nat. Protoc. 2007, 2, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.H.; Venitt, S. DNA and protein adducts in human tissues resulting from exposure to tobacco smoke. Int. J. Cancer 2012, 131, 2733–2753. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, S.M.; Li, H.; Grigoryan, H.; Funk, W.E.; Williams, E.R. Adductomics: Characterizing exposures to reactive electrophiles. Toxicol. Lett. 2012, 213, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.C. 1995 Deichmann Lecture—p53 tumor suppressor gene: At the crossroads of molecular carcinogenesis, molecular epidemiology and cancer risk assessment. Toxicol. Lett. 1995, 82–83, 1–7. [Google Scholar] [CrossRef]
- Wogan, G.N.; Kensler, T.W.; Groopman, J.D. Present and future directions of translational research on aflatoxin and hepatocellular carcinoma. A review. Food Addit. Contam. Part. A Chem. Anal. Control Expo. Risk Assess. 2012, 29, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Stiborova, M.; Schmeiser, H.H. Aristolochic acid as a probable human cancer hazard in herbal remedies: A review. Mutagenesis 2002, 17, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Stiborova, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The role of biotransformation enzymes in the development of renal injury and urothelial cancer caused by aristolochic acid: urgent questions and difficult answers. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2009, 153, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Metabolic activation of carcinogenic aristolochic acid, a risk factor for Balkan endemic nephropathy. Mutat. Res. 2008, 658, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiborová, M.; Frei, E.; Schmeiser, H.H. Biotransformation enzymes in development of renal injury and urothelial cancer caused by aristolochic acid. Kidney Int. 2008, 73, 1209–1211. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Martínek, V.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Enzymes metabolizing aristolochic acid and their contribution to the development of Aristolochic acid nephropathy and urothelial cancer. Curr. Drug Metab. 2013, 14, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Arlt, V.M.; Schmeiser, H.H. Balkan endemic nephropathy: An update on its aetiology. Arch. Toxicol. 2016, 90, 2595–2615. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Stiborova, M.; Arlt, V.M. Chemical and molecular basis of the carcinogenicity of Aristolochia plants. Curr. Opin. Drug Discov. Dev. 2009, 12, 141–148. [Google Scholar]
- Gökmen, M.R.; Cosyns, J.P.; Arlt, V.M.; Stiborová, M.; Phillips, D.H.; Schmeiser, H.H.; Simmonds, M.S.J.; Look, H.T.; Vanherweghem, J.L.; Nortier, J.L.; et al. The epidemiology, diagnosis and management of Aristolochic Acid Nephropathy: A narrative review. Ann. Intern. Med. 2013, 158, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P. Aristolochic acid nephropathy: Harbinger of a global iatrogenic disease. Environ. Mol. Mutagen. 2013, 54, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Pool, B.L.; Wiessler, M. Mutagenicity of the two main components of commercially available carcinogenic aristolochic acid in Salmonella typhimurium. Cancer Lett. 1984, 23, 97–101. [Google Scholar] [CrossRef]
- Kohara, A.; Suzuki, T.; Honma, M.; Ohwada, T.; Hayashi, M. Mutagenicity of aristolochic acid in the lambda/lacZ transgenic mouse (MutaMouse). Mutat. Res. 2002, 515, 63–72. [Google Scholar] [CrossRef]
- Nortier, J.L.; Martinez, M.C.; Schmeiser, H.H.; Arlt, V.M.; Bieler, C.A.; Petein, M.; Depierreux, M.F.; de Pauw, L.; Abramowicz, D.; Vereerstraeten, P.; et al. Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med. 2000, 342, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Mei, N.; Arlt, V.M.; Phillips, D.H.; Heflich, R.H.; Chen, T. DNA adduct formation and mutation induction by aristolochic acid in rat kidney and liver. Mutat. Res. 2006, 602, 83–91. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer (IARC). A review of human CARCINOGENS: Pharmaceuticals. In Environmental Health Criteria Monographs; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Schmeiser, H.H.; Kucab, J.E.; Arlt, V.M.; Phillips, D.H.; Hollstein, M.; Gluhovschi, G.; Gluhovschi, C.; Modilca, M.; Daminescu, L.; Petrica, L.; et al. Evidence of exposure to aristolochic acid in patients with urothelial cancer from a Balkan endemic nephropathy region of Romania. Environ. Mol. Mutagen. 2012, 53, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.H.; Chen, P.C.; Roberts, N.J.; Dickman, K.G.; Yun, B.H.; Turesky, R.J.; Pu, Y.S.; Vogelstein, B.; Papadopoulos, N.; et al. Aristolochic acid in the etiology of renal cell carcinoma. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Rosenquist, T.A.; Grollman, A.P. Mutational signature of aristolochic acid: Clue to the recognition of a global disease. DNA Repair 2016, 44, 205–2011. [Google Scholar] [CrossRef] [PubMed]
- Vanherweghem, J.L.; Tielemans, C.; Abramowicz, D.; Depierreux, M.; Vanhaelen-Fastre, R.; Vanhaelen, M.; Dratwa, M.; Richard, C.; Vandervelde, D.; Verbeelen, D.; et al. Rapidly progressive interstitial renal fibrosis in young women: Association with slimming regimen including Chinese herbs. Lancet 1993, 341, 387–391. [Google Scholar] [CrossRef]
- Arlt, V.M.; Alunni-Perret, V.; Quatrehomme, G.; Ohayon, P.; Albano, L.; Gaïd, H.; Michiels, J.F.; Meyrier, A.; Cassuto, E.; Wiessler, M.; et al. Aristolochic acid (AA)-DNA adduct as marker of AA exposure and risk factor for AA nephropathy-associated cancer. Int. J. Cancer 2004, 111, 977–980. [Google Scholar] [CrossRef] [PubMed]
- Debelle, F.D.; Vanherweghem, J.L.; Nortier, J.L. Aristolochic acid nephropathy: A worldwide problem. Kidney Int. 2008, 74, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Cosyns, J.P.; Goebbels, R.M.; Liberton, V.; Schmeiser, H.H.; Bieler, C.A.; Bernard, A.M. Chinese herbs nephropathy-associated slimming regimen induces tumours in the forestomach but no interstitial nephropathy in rats. Arch. Toxicol. 1998, 72, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Cosyns, J.P.; Jadoul, M.; Squifflet, J.P.; Wese, F.X.; van Ypersele de Strihou, C. Urothelial lesions in Chinese herb nephropathy. Am. J. Kidney Dis. 1999, 33, 1011–1017. [Google Scholar] [CrossRef]
- Jin, K.; Su, K.K.; Li, T.; Zhu, X.Q.; Wang, Q.; Ge, R.S.; Pan, Z.F.; Wu, B.W.; Ge, L.J.; Zhang, Y.H.; et al. Hepatic Premalignant Alterations Triggered by Human Nephrotoxin Aristolochic Acid I in Canines. Cancer Prev. Res. (Phila) 2016, 9, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jin, K.; Zhu, D.Y.; Li, L.; Mao, Z.R.; Wu, B.W.; Wang, Y.F.; Pan, Z.F.; Li, L.J.; Xiang, C.S.; et al. Premalignant alteration assessment in liver-like tissue derived from embryonic stem cells by aristolochic acid I exposure. Oncotarget 2016, 7, 78872–78882. [Google Scholar] [CrossRef] [PubMed]
- Jadot, I.; Declèves, A.E.; Nortier, J.; Caron, N. An integrated view of Aristolochic acid nephropathy: Update of the literature. Int. J. Mol. Sci. 2017, 18, 297. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Pavlović, N.M.; Li, W.; Chan, C.K.; Liu, J.; Deng, K.; Wang, Y.; Milosavljević, B.; Kostić, E.N. Quantitation of Aristolochic Acids in Corn, Wheat Grain, and Soil Samples Collected in Serbia: Identifying a Novel Exposure Pathway in the Etiology of Balkan Endemic Nephropathy. J. Agric. Food Chem. 2016, 64, 5928–5934. [Google Scholar] [CrossRef] [PubMed]
- Pfau, W.; Schmeiser, H.H.; Wiessler, M. Aristolochic acid binds covalently to the exocyclic amino group of purine nucleotides in DNA. Carcinogenesis 1990, 11, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Pfau, W.; Schmeiser, H.H.; Wiessler, M. 32P-postlabelling analysis of the DNA adducts formed by aristolochic acid I and II. Carcinogenesis 1990, 11, 1627–1633. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Bieler, C.A.; Wiessler, M.; van Ypersele de Strihou, C.; Cosyns, J.P. Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res. 1996, 56, 2025–2028. [Google Scholar] [PubMed]
- Schmeiser, H.H.; Frei, E.; Wiessler, M.; Stiborová, M. Comparison of DNA adduct formation by aristolochic acids in various in vitro activation systems by 32P-post-labelling: Evidence for reductive activation by peroxidases. Carcinogenesis 1997, 18, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Fernando, R.C.; Schmeiser, H.H.; Frei, E.; Pfau, W.; Wiessler, M. Characterization of DNA adducts formed by aristolochic acids in the target organ (forestomach) of rats by 32P-postlabelling analysis using different chromatographic procedures. Carcinogenesis 1994, 15, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Mareš, J.; Frei, E.; Arlt, V.M.; Martínek, V.; Schmeiser, H.H. The human carcinogen aristolochic acid I is activated to form DNA adducts by human NAD(P)H:quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen. 2011, 52, 448–459. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Cook, T.; Arlt, V.M.; Schmeiser, H.H.; Williams, G.; Pusey, C.D. Urothelial malignant disease and Chinese herbal nephropathy. Lancet 2001, 358, 1515–1516. [Google Scholar] [CrossRef]
- Lord, G.M.; Hollstein, M.; Arlt, V.M.; Roufosse, C.; Pusey, C.D.; Cook, T.; Schmeiser, H.H. DNA adducts and p53 mutations in a patient with aristolochic acid-associated nephropathy. Am. J. Kidney Dis. 2004, 43, e11–e17. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Stiborova, M.; vom Brocke, J.; Simoes, M.L.; Lord, G.M.; Nortier, J.L.; Hollstein, M.; Phillips, D.H.; Schmeiser, H.H. Aristolochic acid mutagenesis: Molecular clues to the aetiology of Balkan endemic nephropathy-associated urothelial cancer. Carcinogenesis 2007, 28, 2253–2261. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Dekairelle, A.F.; Ambroise, J.; Durant, J.F.; Heusterspreute, M.; Guiot, Y.; Cosyns, J.P.; Gala, J.L. Unambiguous detection of multiple TP53 gene mutations in AAN-associated urothelial cancer in Belgium using laser capture microdissection. PLoS ONE 2014, 9, e106301. [Google Scholar] [CrossRef] [PubMed]
- Aydin, S.; Ambroise, J.; Cosyns, J.P.; Gala, J.L. TP53 mutations in p53-negative dysplastic urothelial cells from Belgian AAN patients: New evidence for aristolochic acid-induced molecular pathogenesis and carcinogenesis. Mutat. Res. 2017, 818, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Nortier, J.L.; Singh, R.; Gamboa da Costa, G.; Sennesael, J.; Cassuto-Viguier, E.; Ambrosetti, D.; Rorive, S.; Pozdzik, A.; Phillips, D.H.; et al. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. Int. J. Cancer 2014, 135, 562–567. [Google Scholar] [CrossRef]
- Schmeiser, H.H.; Janssen, J.W.; Lyons, J.; Scherf, H.R.; Pfau, W.; Buchmann, A.; Bartram, C.R.; Wiessler, M. Aristolochic acid activates ras genes in rat tumors at deoxyadenosine residue. Cancer Res. 1990, 50, 5464–5469. [Google Scholar] [PubMed]
- Schmeiser, H.H.; Scherf, H.R.; Wiessler, M. Activating mutations at codon 61 of the c-Ha-ras gene in thin-tissue sections of tumors induced by aristolochic acid in rats and mice. Cancer Lett. 1991, 59, 139–143. [Google Scholar] [CrossRef]
- Wang, Y.; Meng, F.; Arlt, V.M.; Mei, N.; Chen, T.; Parsons, B.L. Aristolochic acid-induced carcinogenesis examined by ACB-PCR quantification of H-Ras and K-Ras mutant fraction. Mutagenesis 2011, 26, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Arlt, V.M.; Roufosse, C.A.; McKim, K.L.; Myers, M.B.; Phillips, D.H.; Parsons, B.L. ACB-PCR measurement of H-ras codon 61 CAA→CTA mutation provides an early indication of aristolochic acid I carcinogenic effect in tumor target tissues. Environ. Mol. Mutagen. 2012, 53, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Broschard, T.H.; Wiessler, M.; von der Lieth, C.W.; Schmeiser, H.H. Translesional synthesis on DNA templates containing site-specifically placed deoxyadenosine and deoxyguanosine adducts formed by the plant carcinogen aristolochic acid. Carcinogenesis 1994, 15, 2331–2340. [Google Scholar] [CrossRef] [PubMed]
- Moriya, M.; Slade, N.; Brdar, B.; Medverec, Z.; Tomic, K.; Jelakovic, B.; Wu, L.; Truong, S.; Fernandes, A.; Grollman, A.P. TP53 Mutational signature for aristolochic acid: An environmental carcinogen. Int. J. Cancer 2011, 129, 1532–1536. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hollstein, M.; Schmeiser, H.H.; Straif, K.; Wild, C.P. Upper urinary tract urothelial cancer: Where it is A:T. Nat. Rev. Cancer 2012, 12, 503–504. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Pfohl-Leszkowicz, A.; Cosyns, J.; Schmeiser, H.H. Analyses of DNA adducts formed by ochratoxin A and aristolochic acid in patients with Chinese herbs nephropathy. Mutat. Res. 2001, 494, 143–150. [Google Scholar] [CrossRef]
- Bieler, C.A.; Stiborová, M.; Wiessler, M.; Cosyns, J.-P.; van Ypersele de Strihou, C.; Schmeiser, H.H. 32P-postlabelling analysis of DNA adducts formed by aristolochic acid in tissues from patients with Chinese herbs nephropathy. Carcinogenesis 1997, 18, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Ferluga, D.; Stiborova, M.; Pfohl-Leszkowicz, A.; Vukelic, M.; Ceovic, S.; Schmeiser, H.H.; Cosyns, J.P. Is aristolochic acid a risk factor for Balkan endemic nephropathy-associated urothelial cancer? Int. J. Cancer 2002, 101, 500–502. [Google Scholar] [CrossRef] [PubMed]
- Grollman, A.P.; Shibutani, S.; Moriya, M.; Miller, F.; Wu, L.; Moll, U.; Suzuki, N.; Fernandes, A.; Rosenquist, T.; Medverec, Z.; et al. Aristolochic acid and the etiology of endemic Balkan nephropathy. Proc. Natl. Acad. Sci. USA 2007, 104, 12129–12134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jelaković, B.; Karanović, S.; Vuković-Lela, I.; Miller, F.; Edwards, K.L.; Nikolić, J.; Tomić, K.; Slade, N.; Brdar, B.; Turesky, R.J.; et al. Aristolactam-DNA adducts are a biomarker of environmental exposure to aristolochic acid. Kidney Int. 2012, 81, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Rosenquist, T.A.; Sidorenko, V.; Iden, C.R.; Chen, C.H.; Pu, Y.S.; Bonala, R.; Johnson, F.; Dickman, K.G.; Grollman, A.P.; et al. Biomonitoring of aristolactam-DNA adducts in human tissues using ultra-performance liquid chromatography/ion-trap mass spectrometry. Chem. Res. Toxicol. 2012, 25, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Rosenquist, T.A.; Nikolić, J.; Dragičević, D.; Tomić, K.; Jelaković, B.; Dickman, K.G.; Grollman, A.P.; Turesky, R.J. Human formalin-fixed paraffin-embedded tissues: An untapped specimen for biomonitoring of carcinogen DNA adducts by mass spectrometry. Anal. Chem. 2013, 85, 4251–4258. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Sidorenko, V.S.; Rosenquist, T.A.; Dickman, K.G.; Grollman, A.P.; Turesky, R.J. New approaches for biomonitoring exposure to the human carcinogen aristolochic acid. Toxicol. Res. (Camb) 2015, 4, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Stiborova, M.; Arlt, V.M. 32P-postlabeling analysis of DNA adducts. Methods Mol. Biol. 2013, 1044, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Scelo, G.; Riazalhosseini, Y.; Greger, L.; Letourneau, L.; Gonzàlez-Porta, M.; Wozniak, M.B.; Bourgey, M.; Harnden, P.; Egevad, L.; Jackson, S.M.; et al. Variation in genomic landscape of clear cell renal cell carcinoma across Europe. Nat. Commun. 2014, 5, 5135. [Google Scholar] [CrossRef] [PubMed]
- Turesky, R.J.; Yun, B.H.; Brennan, P.; Mates, D.; Jinga, V.; Harnden, P.; Banks, R.E.; Blanche, H.; Bihoreau, M.T.; Chopard, P.; et al. Aristolochic acid exposure in Romania and implications for renal cell carcinoma. Br. J. Cancer 2016, 114, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Dickman, K.G.; Moriya, M.; Zavadil, J.; Sidorenko, V.S.; Edwards, K.L.; Gnatenko, D.V.; Wu, L.; Turesky, R.J.; Wu, X.R.; et al. Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. USA 2012, 109, 8241–8246. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Dickman, K.G.; Huang, C.Y.; Shun, C.T.; Tai, H.C.; Huang, K.H.; Wang, S.M.; Lee, Y.J.; Grollman, A.P.; Pu, Y.S. Recurrence pattern and TP53 mutation in upper urinary tract urothelial carcinoma. Oncotarget 2016, 7, 45225–45236. [Google Scholar] [CrossRef] [PubMed]
- Hoang, M.L.; Chen, C.H.; Sidorenko, V.S.; He, J.; Dickman, K.G.; Yun, B.H.; Moriya, M.; Niknafs, N.; Douville, C.; Karchin, R.; et al. Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med. 2013, 5, 197ra102. [Google Scholar] [CrossRef] [PubMed]
- Poon, S.L.; Pang, S.T.; McPherson, J.R.; Yu, W.; Huang, K.K.; Guan, P.; Weng, W.H.; Siew, E.Y.; Liu, Y.; Heng, H.L.; et al. Genome-wide mutational signatures of aristolochic acid and its application as a screening tool. Sci. Transl. Med. 2013, 5, 197ra101. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Yeo, J.E.; Bonala, R.R.; Johnson, F.; Schärer, O.D.; Grollman, A.P. Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res. 2012, 40, 2494–2505. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Kucab, J.E.; Morganella, S.; Glodzik, D.; Alexandrov, L.B.; Arlt, V.M.; Weninger, A.; Hollstein, M.; Stratton, M.R.; Phillips, D.H. The genome as a record of environmental exposure. Mutagenesis 2015, 30, 763–770. [Google Scholar] [CrossRef] [PubMed]
- National Toxicology Program. Aristolochic Acids 12th Report on Carcinogens; National Toxicology Program; Public Health Service, US Department of Health and Human Services: Research Triangle Park, NC, USA, 2009; pp. 45–49.
- Gillerot, G.; Jadoul, M.; Arlt, V.M.; van Ypersele de Strihou, C.; Schmeiser, H.H.; But, P.P.; Bieler, C.A.; Cosyns, J.P. Aristolochic acid nephropathy in a Chinese patient: Time to abandon the term “Chinese herbs nephropathy”? Am. J. Kidney Dis. 2001, 38, E26. [Google Scholar] [CrossRef] [PubMed]
- Nortier, J.L.; Schmeiser, H.H.; Muniz Martinez, M.C.; Arlt, V.M.; Vervaet, C.; Garbar, C.H.; Daelemans, P.; Vanherweghem, J.L. Invasive urothelial carcinoma after exposure to Chinese herbal medicine containing aristolochic acid may occur without severe renal failure. Nephrol. Dial. Transplant. 2003, 18, 426–428. [Google Scholar] [CrossRef] [PubMed]
- Lo, S.H.; Wong, K.S.; Arlt, V.M.; Phillips, D.H.; Lai, C.K.; Poon, W.T.; Chan, C.K.; Mo, K.L.; Chan, K.W.; Chan, A. Detection of Herba Aristolochia Mollissemae in a patient with unexplained nephropathy. Am. J. Kidney Dis. 2005, 45, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.H.; Yao, L.; Jelaković, B.; Nikolić, J.; Dickman, K.G.; Grollman, A.P.; Rosenquist, T.A.; Turesky, R.J. Formalin-fixed paraffin-embedded tissue as a source for quantitation of carcinogen DNA adducts: Aristolochic acid as a prototype carcinogen. Carcinogenesis 2014, 35, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Roumeguère, T.; Broeders, N.; Jayaswal, A.; Rorive, S.; Quackels, T.; Pozdzik, A.; Arlt, V.M.; Schmeiser, H.H.; Nortier, J.L. Bacillus Calmette-Guerin therapy in non-muscle-invasive bladder carcinoma after renal transplantation for end-stage aristolochic acid nephropathy. Transpl. Int. 2015, 28, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Bamias, G.; Boletis, J. Balkan nephropathy: Evolution of our knowledge. Am. J. Kidney Dis. 2008, 52, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Tatu, C.A.; Orem, W.H.; Finkelman, R.B.; Feder, G.L. The etiology of Balkan endemic nephropathy: Still more questions than answers. Environ. Health Perspect. 1998, 106, 689–700. [Google Scholar] [PubMed]
- Krumbiegel, G.; Hallensleben, J.; Mennicke, W.H.; Rittmann, N. Studies on the metabolism of aristolochic acids I and II. Xenobiotica 1987, 17, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Cu, L.; Xu, G.; Cai, Z. Study of the phase I and phase II metabolism of nephrotoxin aristolochic acid by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006, 20, 1755–1760. [Google Scholar] [CrossRef] [PubMed]
- Chan, W.; Luo, H.B.; Zheng, Y.; Cheng, Y.K.; Cai, Z. Investigation of the metabolism and reductive activation of carcinogenic aristolochic acid in rats. Drug Metab. Dispos. 2007, 35, 866–874. [Google Scholar] [CrossRef] [PubMed]
- Schmeiser, H.H.; Pool, B.L.; Wiessler, M. Identification and mutagenicity of metabolites of aristolochic acid formed by rat liver. Carcinogenesis 1986, 7, 759–763. [Google Scholar] [CrossRef]
- Shibutani, S.; Bonala, R.R.; Rosenquist, T.; Rieger, R.; Suzuki, N.; Johnson, F.; Miller, F.; Grollman, A.P. Detoxification of aristolochic acid I by O-demethylation: Less nephrotoxicity and genotoxicity of aristolochic acid Ia in rodents. Int. J. Cancer 2010, 127, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Suzuki, N.; Torres, M.C.; Bonala, R.R.; Johnson, F.; Grollman, A.P.; Shibutani, S. Quantitative determination of aristolochic acid-derived DNA adducts in rats using 32P-postlabeling/polyacrylamide gel electrophoresis analysis. Drug Metab. Dispos. 2006, 34, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Levová, K.; Bárta, F.; Shi, Z.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Arlt, V.M. Bioactivation versus detoxication of the urothelial carcinogen aristolochic acid I by human cytochrome P450 1A1 and 1A2. Toxicol. Sci. 2012, 125, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Sistkova, J.; Hudecek, J.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Stiborova, M. Human cytochromes P450 1A1 and 1A2 participate in detoxication of carcinogenic aristolochic acid. Neuro Endocrinol. Lett. 2008, 29, 733–737. [Google Scholar] [PubMed]
- Rosenquist, T.A.; Einolf, H.J.; Dickman, K.G.; Wang, L.; Smith, A.; Grollman, A.P. Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos. 2010, 38, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Levová, K.; Mizerovská, M.; Kotrbová, V.; Šulc, M.; Henderson, C.J.; Wolf, C.R.; Philips, D.H.; Frei, E.; Schmeiser, H.H.; Mareš, J.; et al. Role of cytochromes P450 1A1/2 in detoxication and activation of carcinogenic aristolochic acid I: Studies with the hepatic NADPH:cytochrome P450 reductase null (HRN) mouse model. Toxicol. Sci. 2011, 121, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Bárta, F.; Levová, K.; Hodek, P.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. A mechanism of O-demethylation of aristolochic acid I by cytochromes P450 and their contributions to this reaction in human and rat livers: Experimental and theoretical approaches. Int. J. Mol. Sci. 2015, 16, 27561–27575. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Levova, K.; Barta, F.; Shi, Z.; Evans, J.D.; Frei, E.; Schmeiser, H.H.; Nebert, D.W.; Phillips, D.H.; Stiborova, M. Role of P450 1A1 and P450 1A2 in bioactivation versus detoxication of the renal carcinogen aristolochic acid I: Studies in Cyp1a1(−/−), Cyp1a2(−/−), and Cyp1a1/1a2(−/−) mice. Chem. Res. Toxicol. 2011, 24, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Dračínská, H.; Bárta, F.; Levová, K.; Hudecová, A.; Moserová, M.; Schmeiser, H.H.; Kopka, K.; Frei, E.; Arlt, V.M.; Stiborová, M. Induction of cytochromes P450 1A1 and 1A2 suppresses formation of DNA adducts by carcinogenic aristolochic acid I in rats in vivo. Toxicology 2016, 344, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Xiao, Y.; Zhu, H.; Wang, H.; Liu, Y.; Xie, T.; Ren, J. Induction of P450 1A by 3-methylcholanthrene protects mice from aristolochic acid-I-induced acute renal injury. Nephrol. Dial. Transplant. 2008, 23, 3074–3081. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Ge, M.; Xue, X.; Wang, H.; Wu, X.; Li, L.; Liu, L.; Qi, X.; Zhang, Y.; Li, Y.; et al. Detoxication role of hepatic cytochrome P450s in the kidney toxicity induced by aristolochic acid. Kidney Int. 2008, 73, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Martínek, V.; Bárta, F.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. Comparison of the oxidation of carcinogenic aristolochic acid I and II by microsomal cytochromes P450 in vitro: Experimental and theoretical approaches. Monatshefte Chem. 2017. [Google Scholar]
- Stiborova, M.; Barta, F.; Dracinska, H.; Hudecova, A.; Hodek, P.; Balogova, M.; Mraz, J.; Duskova, S.; Schmeiser, H.H.; Arlt, V.M. Treatment with a mixture of aristolochic acid I and II influences their genotoxicity and expression of biotransformation enzymes in rats in vivo. Toxicol. Lett. 2016, 256, S96. [Google Scholar] [CrossRef]
- Schmeiser, H.; Schoepe, K.B.; Wiessler, M. DNA adduct formation of aristolochic acid I and II in vitro and in vivo. Carcinogenesis 1988, 9, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Shibutani, S.; Dong, H.; Suzuki, N.; Ueda, S.; Miller, F.; Grollman, A.P. Selective toxicity of aristolochic acids I and II. Drug Metab. Dispos. 2007, 35, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Arlt, V.M.; Meinl, W.; Florian, S.; Nagy, E.; Barta, F.; Thomann, M.; Mrizova, I.; Krais, A.M.; Liu, M.; Richards, M.; et al. Impact of genetic modulation of SULT1A enzymes on DNA adduct formation by aristolochic acids and 3-nitrobenzanthrone. Arch. Toxicol. 2017, 91, 1957–1975. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Wiessler, M.; Schmeiser, H.H. Human enzymes involved in the metabolic activation of carcinogenic aristolochic acids: Evidence for reductive activation by cytochromes P450 1A1 and 1A2. Chem. Res. Toxicol. 2001, 14, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Hájek, M.; Frei, E.; Schmeiser, H.H. Carcinogenic and nephrotoxic alkaloids aristolochic acids upon activation by NADPH:cytochrome P450 reductase form adducts found in DNA of patients with Chinese herbs nephropathy. Gen. Physiol. Biophys. 2001, 20, 375–392. [Google Scholar] [PubMed]
- Stiborová, M.; Frei, E.; Breuer, A.; Wiessler, M.; Schmeiser, H.H. Evidence for reductive activation of carcinogenic aristolochic acids by prostaglandin H synthase—32P-postlabeling analysis of DNA adduct formation. Mutat. Res. 2001, 493, 149–160. [Google Scholar] [CrossRef]
- Stiborová, M.; Frei, E.; Sopko, B.; Wiessler, M.; Schmeiser, H.H. Carcinogenic aristolochic acids upon activation by DT-diaphorase form adducts found in DNA of patients with Chinese herbs nephropathy. Carcinogenesis 2002, 23, 617–625. [Google Scholar] [CrossRef] [PubMed]
- Martínek, V.; Kubickova, B.; Arlt, V.M.; Frei, E.; Schmeiser, H.H.; Hudeček, J.; Stiborova, M. Comparison of activation of aristolochic acid I and II with NADPH:quinone oxidoreductase, sulphotransferases and N-acetyltransferases. Neuro Endocrinol. Lett. 2011, 32 (Suppl. S1), S57–S70. [Google Scholar]
- Stiborová, M.; Hudeček, J.; Frei, E.; Schmeiser, H.H. Contribution of biotransformation enzymes to the development of renal injury and urothelial cancer caused by aristolochic acid: Urgent questions, difficult answers. Interdiscip. Toxicol. 2008, 1, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Sopko, B.; Sopková, K.; Marková, V.; Laňková, M.; Kumstýřová, T.; Wiessler, M.; Schmeiser, H.H. Human cytosolic enzymes involved in the metabolic activation of carcinogenic aristolochic acid: Evidence for reductive activation by human NAD(P)H:quinone oxidoreductase. Carcinogenesis 2003, 24, 1695–1703. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Gong, L.; Qi, X.; Xing, G.; Luan, Y.; Wu, Y.; Xiao, Y.; Yao, J.; Li, Y.; Xue, X.; et al. Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci. 2011, 122, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. Knock-out and humanized mice as suitable tools to identify enzymes metabolizing the human carcinogen aristolochic acid. Xenobiotica 2014, 44, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Martínek, V. Mechanisms of enzyme-catalyzed reduction of two carcinogenic nitro-aromatics, 3-nitrobenzanthrone and aristolochic acid I: Experimental and theoretical approaches. Int. J. Mol. Sci. 2014, 15, 10271–10295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiborová, M.; Levová, K.; Bárta, F.; Šulc, M.; Frei, E.; Arlt, V.M.; Schmeiser, H.H. The influence of dicoumarol on the bioactivation of the carcinogen aristolochic acid I in rats. Mutagenesis 2014, 29, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Attaluri, S.; Zaitseva, I.; Iden, C.R.; Dickman, K.G.; Johnson, F.; Grollman, A.P. Bioactivation of the human carcinogen aristolochic acid. Carcinogenesis 2014, 35, 1814–1822. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Zaitseva, I.N.; Bonala, R.; Attaluri, S.; Ozga, K.; Iden, C.R.; Johnson, F.; Moriya, M.; Grollman, A.P.; Sidorenko, V.S. Sulfotransferase-1A1-dependent bioactivation of aristolochic acid I and N-hydroxyaristolactam I in human cells. Carcinogenesis 2016, 37, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Meinl, W.; Pabel, U.; Osterloh-Quiroz, M.; Hengstler, J.G.; Glatt, H. Human sulphotransferases are involved in the activation of aristolochic acids and are expressed in renal target tissue. Int. J. Cancer 2006, 118, 1090–1097. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Frei, E.; Hodek, P.; Wiessler, M.; Schmeiser, H.H. Human hepatic and renal microsomes, cytochromes P450 1A1/2, NADPH:CYP reductase and prostaglandin H synthase mediate the formation of aristolochic acid DNA-adducts found in patients with urothelial cancer. Int. J. Cancer 2005, 113, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Stiborová, M.; Sopko, B.; Hodek, P.; Frei, E.; Schmeiser, H.H.; Hudeček, J. The binding of aristolochic acid I to the active site of human cytochromes P450 1A1 and 1A2 explains their potential to reductively activate this human carcinogen. Cancer Lett. 2005, 229, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Milichovský, J.; Bárta, F.; Schmeiser, H.H.; Arlt, V.M.; Frei, E.; Stiborová, M.; Martínek, V. Active site mutations as a suitable tool contributing to explain a mechanism of aristolochic acid I nitroreduction by cytochromes P450 1A1, 1A2, and 1B1. Int. J. Mol. Sci. 2016, 7, 213. [Google Scholar] [CrossRef]
- Jerabek, P.; Martinek, V.; Stiborova, M. Theoretical investigation of differences in nitroreduction of aristolochic acid I by cytochromes P450 1A1, 1A2 and 1B1. Neuro Endocrinol. Lett. 2012, 33 (Suppl. S3), 25–32. [Google Scholar] [PubMed]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Cytochrome P450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Rendic, S.; DiCarlo, F.J. Human cytochrome P450 enzymes: A status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab. Rev. 1997, 29, 413–480. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, S.Y.; von Ahsen, N.; Toncheva, D.I.; Dimitrov, T.G.; Oellerich, M.; Amstrong, V.M. Genetic polymorphism of cytochrome P450 among patients with Balkan endemic nephropathy (BEN). Clin. Biochem. 2005, 38, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Kepa, J.K.; Winski, S.L.; Beall, H.D.; Anwar, A.; Siegel, D. NAD(P)H:quinone oxidoreductase 1 (NQO1): Chemoprotection, bioactivation, gene regulation and genetic polymorphisms. Chem. Biol. Interact. 2000, 129, 77–97. [Google Scholar] [CrossRef]
- Ross, D. Quinone reductases multitasking in the metabolic world. Drug Metab. Rev. 2004, 36, 639–654. [Google Scholar] [CrossRef] [PubMed]
- Bárta, F.; Levová, K.; Frei, E.; Schmeiser, H.H.; Arlt, V.M.; Stiborová, M. The effect of aristolochic acid I on NAD(P)H:quinone oxidoreductase expression in mice and rats—A comparative study. Mutat. Res. 2014, 768, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toncheva, D.; Dimitrov, T.; Stojanova, S. Etiology of Balkan endemic nephropathy: A multifactorial disease? Eur. J. Epidemiol. 1998, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, D.I.; von Ahsen, N.; Atanasova, S.Y.; Dimitrov, T.G.; Armstrong, V.M.; Oellerich, M. Identification of NQO1 and GSTs genotype frequencies in Bulgarian patients with Balkan endemic nephropathy. J. Nephrol. 2004, 17, 384–389. [Google Scholar] [PubMed]
- He, P.; Court, R.H.; Greenblatt, D.J.; Von Moltke, L.L. Genotype-phenotype associations of cytochrome P450 3A4 and 3A5 polymorphism with midazolam clearance in vivo. Clin. Pharmacol. Ther. 2005, 77, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Stefanović, V.; Toncheva, D.; Atanasova, S.; Polenaković, M. Etiology of Balkan endemic nephropathy and associated urothelial cancer. Am. J. Nephrol. 2006, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Toncheva, D. Genetic studies in BEN and associated urothelial cancers. Coll. Antropol. 2006, 30 (Suppl. S1), 34. [Google Scholar]
- Chen, B.; Bai, Y.; Sun, M.; Ni, X.; Yang, Y.; Yang, Y.; Zheng, S.; Xu, F.; Dai, S. Glutathione S-transferases T1 null genotype is associated with susceptibility to aristolochic acid nephropathy. Int. Urol. Nephrol. 2012, 44, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Huang, S.Q.; Su, H.H.; Zhou, S.F. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr. Drug Metab. 2009, 10, 781–834. [Google Scholar] [CrossRef] [PubMed]
- Reljic, Z.; Zlatovic, M.; Savic-Radojevic, A.; Pekmezovic, T.; Djukanovic, L.; Matic, M.; Pljesa-Ercegovac, M.; Mimic-Oka, J.; Opsenica, D.; Simic, T. Is increased susceptibility to Balkan endemic nephropathy in carriers of common GSTA1 (*A/*B) polymorphism linked with the catalytic role of GSTA1 in ochratoxin a biotransformation? Serbian case control study and in silico analysis. Toxins 2014, 6, 2348–2362. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | DNA Adduct Measured in | DNA Adduct Detected | DNA Adduct Level/108 Nucleotides | Method Used for DNA Adduct Detection | Publication Year | Reference |
|---|---|---|---|---|---|---|
| Belgium | Kidney | dA-AAI | 7.0–53.0 (n = 6) | TLC 1 32P-postlabelling | 1996, 1997 | [44,62] |
| Belgium | Kidney | dA-AAI | 0.1–16.5 (n = 38) | TLC 32P-postlabelling | 2000 | [27] |
| Belgium | Kidney | dA-AAI | 2.9–5.0 (n = 2) | TLC 32P-postlabelling | 2001 | [61] |
| Belgium/China | Kidney | dA-AAI | 1.8 (n = 1) | TLC 32P-postlabelling | 2001 | [79] |
| UK | Kidney | dA-AAI | 3.8 (n = 1) | TLC 32P-postlabelling | 2001, 2004 | [48,49] |
| Croatia | Kidney | dA-AAI | 0.56–1.71 (n = 2) | TLC 32P-postlabelling | 2002 | [63] |
| Belgium | Kidney | dA-AAI | 8.1 (n = 1) | TLC 32P-postlabelling | 2003 | [80] |
| France | Kidney | dA-AAI | 0.1–5.4 (n = 2) | TLC 32P-postlabelling | 2004 | [34] |
| China | Kidney | dA-AAI | Detected, but not quantified | TLC 32P-postlabelling | 2005 | [81] |
| USA | Kidney (cortex, medulla, and pelvis) Kidney (cortex) | dA-AA dA-AAI | 11.0–34 (n = 1) Detected, but not quantified (n = 1) | PAGE 2 32P-postlabelling Mass spectrometry | 2007 | [64] |
| Croatia | Kidney | dA-AA | 8–59 (n = 4) | PAGE 32P-postlabelling | 2007 | [64] |
| Taiwan | Kidney | dA-AA | 1.4–234 (89/148 (60%)) | PAGE 32P-postlabelling | 2012 | [72] |
| Bosnia, Croatia & Serbia | Kidney | dA-AA | 0.2–19.2 (47/67 (70%)) | PAGE 32P-postlabelling | 2012 | [65] |
| Romania | Kidney | dA-AAI | 0.3–6.5 (n = 7) | TLC 32P-postlabelling | 2012 | [30] |
| Belgium | Kidney Kidney | dA-AAI dA-AAI | 2–22 (n = 11) Detected, but not quantified (n = 1) | TLC 32P-postlabelling Mass spectrometry | 2014 | [53] |
| Croatia & Serbia | Kidney | dA-AAI | 0.2–7.0 (n = 15) | Mass spectrometry | 2014 | [82] |
| Belgium | Kidney | dA-AAI | 5 (n = 1) | TLC 32P-postlabelling | 2015 | [83] |
| Romania | Kidney | dA-AAI | 0.7–26.8 (n = 14) | Mass spectrometry | 2016 | [71] |
| Taiwan | Kidney | dA-AAI | 0.3–258 (39/51 (76%)) | Mass spectrometry | 2016 | [31] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiborová, M.; Arlt, V.M.; Schmeiser, H.H. DNA Adducts Formed by Aristolochic Acid Are Unique Biomarkers of Exposure and Explain the Initiation Phase of Upper Urothelial Cancer. Int. J. Mol. Sci. 2017, 18, 2144. https://doi.org/10.3390/ijms18102144
Stiborová M, Arlt VM, Schmeiser HH. DNA Adducts Formed by Aristolochic Acid Are Unique Biomarkers of Exposure and Explain the Initiation Phase of Upper Urothelial Cancer. International Journal of Molecular Sciences. 2017; 18(10):2144. https://doi.org/10.3390/ijms18102144
Chicago/Turabian StyleStiborová, Marie, Volker M. Arlt, and Heinz H. Schmeiser. 2017. "DNA Adducts Formed by Aristolochic Acid Are Unique Biomarkers of Exposure and Explain the Initiation Phase of Upper Urothelial Cancer" International Journal of Molecular Sciences 18, no. 10: 2144. https://doi.org/10.3390/ijms18102144
APA StyleStiborová, M., Arlt, V. M., & Schmeiser, H. H. (2017). DNA Adducts Formed by Aristolochic Acid Are Unique Biomarkers of Exposure and Explain the Initiation Phase of Upper Urothelial Cancer. International Journal of Molecular Sciences, 18(10), 2144. https://doi.org/10.3390/ijms18102144