Redox Signaling and Its Impact on Skeletal and Vascular Responses to Spaceflight

{kind=link}
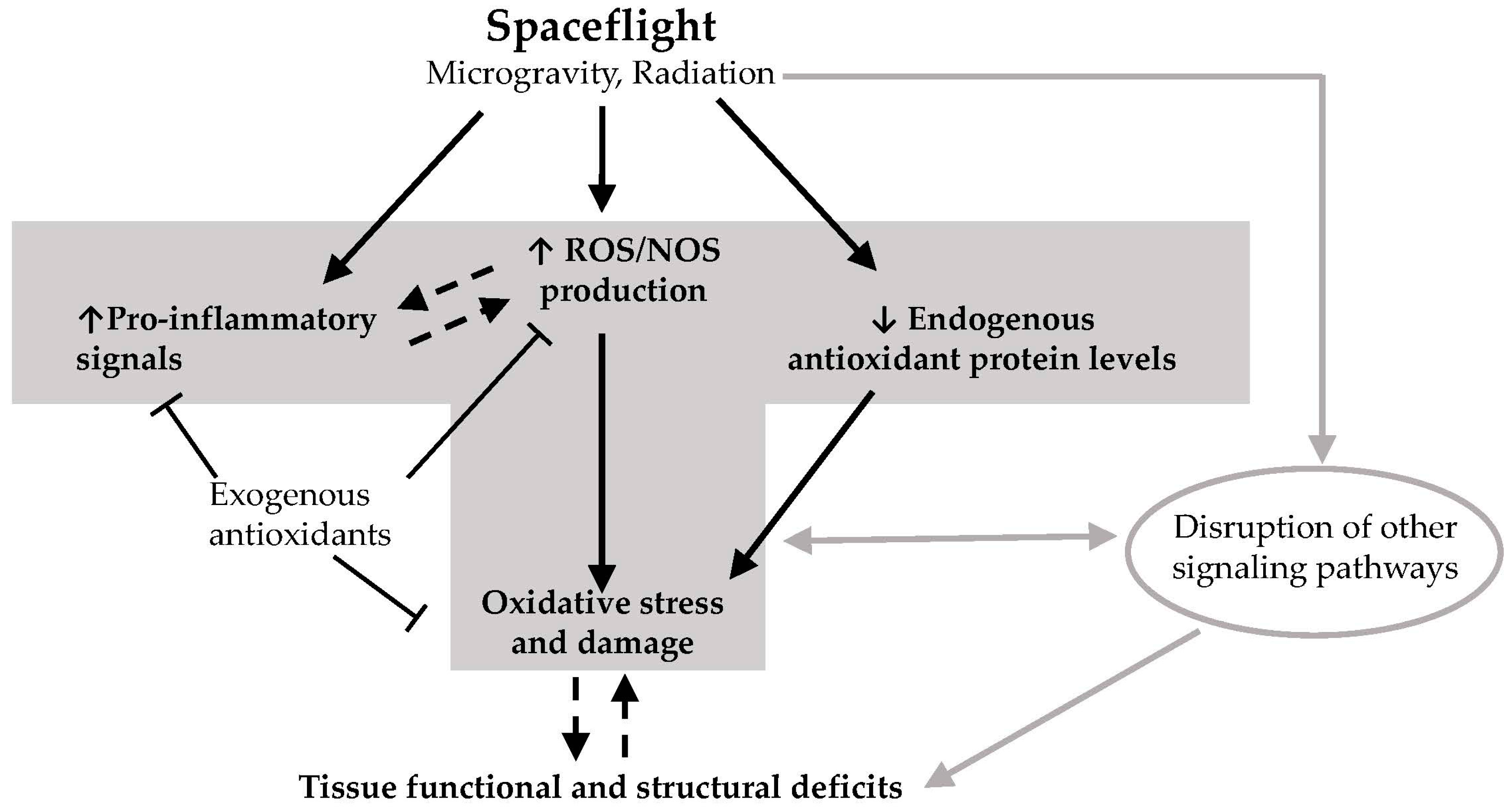
Abstract
:1. The Spaceflight Environment and Its Impact on Skeletal and Vascular Health
2. Oxidative Stress and Its Link to Spaceflight-Induced Tissue Dysfunction
2.1. Oxidative Damage Associated with Spaceflight and Its Analogs
2.1.1. Evidence from Spaceflight
2.1.2. Evidence from Ground-Based Models for Spaceflight
3. The Role of Nitric Oxide (NO) and Reactive Oxygen Species (ROS) Signaling in Skeletal and Vascular Disease
3.1. NO and ROS Signaling: Mechanisms and Impact on Tissue Function
3.2. Bone and Vascular Function during Development Are Intimately Associated
3.3. Vascular-Bone Coupling Occurs via Redox-Dependent Mechanisms: Implications for Tissue Responses to Spaceflight
3.4. Cellular Defenses to Oxidative Damage Are Important for Preserving Skeletal and Vascular Health
3.4.1. Nuclear Factor Erythroid 2-Related Factor 2 (NRF2)
3.4.2. CuZn Superoxide Dismutase (SOD1)
3.4.3. Catalase
4. Implications for the Development of Spaceflight Countermeasures
5. Concluding Statements
Acknowledgments
Conflicts of Interest
References
- Lang, T.; LeBlanc, A.; Evans, H.; Lu, Y.; Genant, H.; Yu, A. Cortical and trabecular bone mineral loss from the spine and hip in long-duration spaceflight. J. Bone Miner. Res. 2004, 19, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, A.; Schneider, V.; Shackelford, L.; West, S.; Oganov, V.; Bakulin, A.; Voronin, L. Bone mineral and lean tissue loss after long duration space flight. J. Musculoskelet. Neuronal Interact. 2000, 1, 157–176. [Google Scholar] [PubMed]
- Sibonga, J.D.; Evans, H.J.; Sung, H.G.; Spector, E.R.; Lang, T.F.; Oganov, V.S.; Bakulin, A.V.; Shackelford, L.C.; LeBlanc, A.D. Recovery of spaceflight-induced bone loss: Bone mineral density after long-duration missions as fitted with an exponential function. Bone 2007, 41, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Keyak, J.H.; Koyama, A.K.; LeBlanc, A.; Lu, Y.; Lang, T.F. Reduction in proximal femoral strength due to long-duration spaceflight. Bone 2009, 44, 449–495. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, S.A.; Hogan, H.A.; Delp, M.D. Decreases in bone blood flow and bone material properties in aging Fischer-344 rats. Clin. Orthop. Relat. Res. 2002, 248–285. [Google Scholar] [CrossRef]
- Prisby, R.D.; Ramsey, M.W.; Behnke, B.J.; Dominguez, J.M., 2nd; Donato, A.J.; Allen, M.R.; Delp, M.D. Aging reduces skeletal blood flow, endothelium-dependent vasodilation, and NO bioavailability in rats. J. Bone Miner. Res. 2007, 22, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, R.; Kettner, G.; Bohm, W.; Schmidmeier, M.; Schlag, R.; Frisch, B.; Mallmann, B.; Eisenmenger, W.; Gilg, T. Changes in trabecular bone, hematopoiesis and bone marrow vessels in aplastic anemia, primary osteoporosis, and old age: A comparative histomorphometric study. Bone 1987, 8, 157–176. [Google Scholar] [CrossRef] [Green Version]
- National Council on Radiation Protection and Measurements (NCRP). Information Needed to Make Recommendations for Space Missions Beyond Low-Earth Orbit; US National Council for Radiation Protection and Measurements: Bethesda, MD, USA, 2006. [Google Scholar]
- Chancellor, J.C.; Scott, G.B.; Sutton, J.P. Space radiation: The number one risk to astronaut health beyond low earth orbit. Life 2014, 4, 491–510. [Google Scholar] [CrossRef] [PubMed]
- Silberberg, R.; Tsao, C.H.; Adams, J.H., Jr.; Letaw, J.R. Radiation doses and LET distributions of cosmic rays. Radiat. Res. 1984, 98, 209–292. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.A.; Katz, R.; Wilson, J.W.; Townsend, L.W.; Shinn, J.; Hajnal, F. Biological effectiveness of high-energy protons: Target fragmentation. Radiat. Res. 1991, 127, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.W.; Townsend, L.W.; Badavi, F.F. Galactic HZE propagation through the Earth’s atmosphere. Radiat. Res. 1987, 109, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, D.M.; Asaithamby, A.; Bailey, S.M.; Costes, S.V.; Doetsch, P.W.; Dynan, W.S.; Kronenberg, A.; Rithidech, K.N.; Saha, J.; Snijders, A.M.; et al. Understanding cancer development processes after HZE-particle exposure: Roles of ROS, DNA damage repair and inflammation. Radiat. Res. 2015, 183, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Saha, J.; Wilson, P.; Thieberger, P.; Lowenstein, D.; Wang, M.; Cucinotta, F.A. Biological characterization of low-energy ions with high-energy deposition on human cells. Radiat. Res. 2014, 182, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Limoli, C.L.; Giedzinski, E.; Rola, R.; Otsuka, S.; Palmer, T.D.; Fike, J.R. Radiation response of neural precursor cells: Linking cellular sensitivity to cell cycle checkpoints, apoptosis and oxidative stress. Radiat. Res. 2004, 161, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed]
- Mavragani, I.V.; Nikitaki, Z.; Souli, M.P.; Aziz, A.; Nowsheen, S.; Aziz, K.; Rogakou, E.; Georgakilas, A.G. Complex DNA Damage: A Route to Radiation-Induced Genomic Instability and Carcinogenesis. Cancers 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.; Gore, M.; Fiebig, R.; Mazzeo, R.; Ohishi, S.; Ohno, H.; Ji, L.L. Spaceflight downregulates antioxidant defense systems in rat liver. Free Radic. Biol. Med. 1998, 24, 385–390. [Google Scholar] [CrossRef]
- Stein, T.P.; Leskiw, M.J. Oxidant damage during and after spaceflight. Am. J. Physiol. Endocrinol. Metab. 2000, 278, E375–E382. [Google Scholar] [PubMed]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar] [CrossRef] [PubMed]
- De Luca, C.; Deeva, I.; Mariani, S.; Maiani, G.; Stancato, A.; Korkina, L. Monitoring antioxidant defenses and free radical production in space-flight, aviation and railway engine operators, for the prevention and treatment of oxidative stress, immunological impairment, and pre-mature cell aging. Toxicol. Ind. Health 2009, 25, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Davis-Street, J.E.; Rice, B.L.; Nillen, J.L.; Gillman, P.L.; Block, G. Nutritional status assessment in semiclosed environments: Ground-based and space flight studies in humans. J. Nutr. 2001, 131, 2053–2061. [Google Scholar] [PubMed]
- Smith, S.M.; Zwart, S.R.; Block, G.; Rice, B.L.; Davis-Street, J.E. The nutritional status of astronauts is altered after long-term space flight aboard the International Space Station. J. Nutr. 2005, 135, 437–474. [Google Scholar] [PubMed]
- Indo, H.P.; Majima, H.J.; Terada, M.; Suenaga, S.; Tomita, K.; Yamada, S.; Higashibata, A.; Ishioka, N.; Kanekura, T.; Nonaka, I.; et al. Changes in mitochondrial homeostasis and redox status in astronauts following long stays in space. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Connor, M.K.; Hood, D.A. Effect of microgravity on the expression of mitochondrial enzymes in rat cardiac and skeletal muscles. J. Appl. Physiol. 1998, 84, 593–598. [Google Scholar] [PubMed]
- Ray, C.A.; Vasques, M.; Miller, T.A.; Wilkerson, M.K.; Delp, M.D. Effect of short-term microgravity and long-term hindlimb unloading on rat cardiac mass and function. J. Appl. Physiol. 2001, 91, 1207–1213. [Google Scholar] [PubMed]
- Taylor, C.R.; Hanna, M.; Behnke, B.J.; Stabley, J.N.; McCullough, D.J.; Davis, R.T., 3rd; Ghosh, P.; Papadopoulos, A.; Muller-Delp, J.M.; Delp, M.D. Spaceflight-induced alterations in cerebral artery vasoconstrictor, mechanical, and structural properties: Implications for elevated cerebral perfusion and intracranial pressure. FASEB J. 2013, 27, 2282–2292. [Google Scholar] [CrossRef] [PubMed]
- Dabertrand, F.; Porte, Y.; Macrez, N.; Morel, J.L. Spaceflight regulates ryanodine receptor subtype 1 in portal vein myocytes in the opposite way of hypertension. J. Appl. Physiol. 2012, 112, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Baqai, F.P.; Gridley, D.S.; Slater, J.M.; Luo-Owen, X.; Stodieck, L.S.; Ferguson, V.; Chapes, S.K.; Pecaut, M.J. Effects of spaceflight on innate immune function and antioxidant gene expression. J. Appl. Physiol. 2009, 106, 1935–1942. [Google Scholar] [CrossRef] [PubMed]
- Mader, T.H.; Gibson, C.R.; Pass, A.F.; Kramer, L.A.; Lee, A.G.; Fogarty, J.; Tarver, W.J.; Dervay, J.P.; Hamilton, D.R.; Sargsyan, A.; et al. Optic disc edema, globe flattening, choroidal folds, and hyperopic shifts observed in astronauts after long-duration space flight. Ophthalmology 2011, 118, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.W.; Pecaut, M.J.; Stodieck, L.S.; Ferguson, V.L.; Bateman, T.A.; Bouxsein, M.; Jones, T.A.; Moldovan, M.; Cunningham, C.E.; Chieu, J.; et al. Spaceflight environment induces mitochondrial oxidative damage in ocular tissue. Radiat. Res. 2013, 180, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Sasi, S.P.; Gee, H.; Lee, J.; Yang, Y.; Mehrzad, R.; Onufrak, J.; Song, J.; Enderling, H.; Agarwal, A.; et al. Cardiovascular risks associated with low dose ionizing particle radiation. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Kondo, H.; Yumoto, K.; Alwood, J.S.; Mojarrab, R.; Wang, A.; Almeida, E.A.; Searby, N.D.; Limoli, C.L.; Globus, R.K. Oxidative stress and γ radiation-induced cancellous bone loss with musculoskeletal disuse. J. Appl. Physiol. 2010, 108, 126–152. [Google Scholar] [CrossRef] [PubMed]
- Alwood, J.S.; Shahnazari, M.; Chicana, B.; Schreurs, A.S.; Kumar, A.; Bartolini, A.; Shirazi-Fard, Y.; Globus, R.K. Ionizing radiation stimulates expression of pro-osteoclastogenic genes in marrow and skeletal tissue. J. Interferon Cytokine Res. 2015, 35, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Schreurs, A.S.; Shirazi-Fard, Y.; Shahnazari, M.; Alwood, J.S.; Truong, T.A.; Tahimic, C.G.; Limoli, C.L.; Turner, N.D.; Halloran, B.; Globus, R.K. Dried plum diet protects from bone loss caused by ionizing radiation. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Morey-Holton, E.R.; Globus, R.K. Hindlimb unloading rodent model: Technical aspects. J. Appl. Physiol. 2002, 92, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Globus, R.K.; Morey-Holton, E. Hindlimb unloading: Rodent analog for microgravity. J. Appl. Physiol. 2016, 120, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, S.A.; Martinez, D.A.; Boudreaux, R.D.; Mantri, A.V. Microgravity stress: Bone and connective tissue. Compr. Physiol. 2016, 6, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, D.; Nojiri, H.; Saita, Y.; Kobayashi, K.; Watanabe, K.; Ozawa, Y.; Koike, M.; Asou, Y.; Takaku, T.; Kaneko, K.; et al. Cytoplasmic reactive oxygen species and SOD1 regulate bone mass during mechanical unloading. J. Bone Miner. Res. 2013, 28, 2368–2380. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.A.; Henry, M.K.; Welliver, K.C.; Jepson, A.J.; Garnett, E.R. Hindlimb unloading results in increased predisposition to cardiac arrhythmias and alters left ventricular connexin 43 expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R362–R373. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Fan, Y.; Sun, A.; Jia, X.; Deng, X. Simulated microgravity exposure modulates the phenotype of cultured vascular smooth muscle cells. Cell Biochem. Biophys. 2013, 66, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Tsvirkun, D.; Bourreau, J.; Mieuset, A.; Garo, F.; Vinogradova, O.; Larina, I.; Navasiolava, N.; Gauquelin-Koch, G.; Gharib, C.; Custaud, M.A. Contribution of social isolation, restraint, and hindlimb unloading to changes in hemodynamic parameters and motion activity in rats. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, A.S.; Harrison, R.; Lee, K.H.; Genut, J.; Nyhan, D.; Brooks-Asplund, E.M.; Shoukas, A.A.; Hare, J.M.; Berkowitz, D.E. Simulated microgravity produces attenuated baroreflex-mediated pressor, chronotropic, and inotropic responses in mice. Am. J. Physiol Heart Circ. Physiol. 2005, 289, H600–H607. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.; Bernstein, D. The mouse as a model of cardiovascular adaptations to microgravity. J. Appl. Physiol. 2004, 97, 1686–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Bai, Y.G.; Lin, L.J.; Bao, J.X.; Zhang, Y.Y.; Tang, H.; Cheng, J.H.; Jia, G.L.; Ren, X.L.; Ma, J. Blockade of AT1 receptor partially restores vasoreactivity, NOS expression, and superoxide levels in cerebral and carotid arteries of hindlimb unweighting rats. J. Appl. Physiol. 2009, 106, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ran, H.H.; Ma, J.; Bai, Y.G.; Lin, L.J. NAD(P)H oxidase inhibiting with apocynin improved vascular reactivity in tail-suspended hindlimb unweighting rat. J. Physiol. Biochem. 2012, 68, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Kanazashi, M.; Okumura, Y.; Al-Nassan, S.; Murakami, S.; Kondo, H.; Nagatomo, F.; Fujita, N.; Ishihara, A.; Roy, R.R.; Fujino, H. Protective effects of astaxanthin on capillary regression in atrophied soleus muscle of rats. Acta Physiol. 2013, 207, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Lean, J.M.; Davies, J.T.; Fuller, K.; Jagger, C.J.; Kirstein, B.; Partington, G.A.; Urry, Z.L.; Chambers, T.J. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J. Clin. Investig. 2003, 112, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Garrett, I.R.; Boyce, B.F.; Oreffo, R.O.; Bonewald, L.; Poser, J.; Mundy, G.R. Oxygen-derived free radicals stimulate osteoclastic bone resorption in rodent bone in vitro and in vivo. J. Clin. Investig. 1990, 85, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Han, L.; Martin-Millan, M.; Plotkin, L.I.; Stewart, S.A.; Roberson, P.K.; Kousteni, S.; O’Brien, C.A.; Bellido, T.; Parfitt, A.M.; et al. Skeletal involution by age-associated oxidative stress and its acceleration by loss of sex steroids. J. Biol. Chem. 2007, 282, 27285–27297. [Google Scholar] [CrossRef] [PubMed]
- Manolagas, S.C.; Almeida, M. Gone with the Wnts: β-Catenin, T-cell factor, forkhead box O, and oxidative stress in age-dependent diseases of bone, lipid, and glucose metabolism. Mol. Endocrinol. 2007, 21, 2605–2614. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Lee, J.H.; Kim, H.N.; Kim, H.M.; Kwak, H.B.; Lee, S.; Kim, H.H.; Lee, Z.H. α-Lipoic acid inhibits inflammatory bone resorption by suppressing prostaglandin E2 synthesis. J. Immunol. 2006, 176, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Fisher, A.B. Mechanotransduction: Forces, sensors, and redox signaling. Antioxid. Redox Signal. 2014, 20, 868–887. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.J.; Liu, C.A.; Huang, B.; Tseng, A.H.; Wang, D.L. Shear-induced endothelial mechanotransduction: The interplay between reactive oxygen species (ROS) and nitric oxide (NO) and the pathophysiological implications. J. Biomed. Sci. 2014, 21. [Google Scholar] [CrossRef] [PubMed]
- Noris, M.; Morigi, M.; Donadelli, R.; Aiello, S.; Foppolo, M.; Todeschini, M.; Orisio, S.; Remuzzi, G.; Remuzzi, A. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ. Res. 1995, 76, 536–564. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, Y.; Ohhashi, T. Acetylcholine- and flow-induced production and release of nitric oxide in arterial and venous endothelial cells. Am. J. Physiol. 1996, 270, H99–H106. [Google Scholar] [PubMed]
- Korenaga, R.; Ando, J.; Tsuboi, H.; Yang, W.; Sakuma, I.; Toyo-oka, T.; Kamiya, A. Laminar flow stimulates ATP- and shear stress-dependent nitric oxide production in cultured bovine endothelial cells. Biochem. Biophys. Res. Commun. 1994, 198, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Klein-Nulend, J.; Helfrich, M.H.; Sterck, J.G.; MacPherson, H.; Joldersma, M.; Ralston, S.H.; Semeins, C.M.; Burger, E.H. Nitric oxide response to shear stress by human bone cell cultures is endothelial nitric oxide synthase dependent. Biochem. Biophys. Res. Commun. 1998, 250, 108–181. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.D.; Soejima, K.; Klein-Nulend, J.; Burger, E.H. The production of nitric oxide and prostaglandin E(2) by primary bone cells is shear stress dependent. J. Biomech. 2001, 34, 671–677. [Google Scholar] [CrossRef]
- Godo, S.; Shimokawa, H. Divergent roles of endothelial nitric oxide synthases system in maintaining cardiovascular homeostasis. Free Radic. Biol. Med. 2017, 109, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Lotem, J.; Peled-Kamar, M.; Groner, Y.; Sachs, L. Cellular oxidative stress and the control of apoptosis by wild-type p53, cytotoxic compounds, and cytokines. Proc. Natl. Acad. Sci. USA 1996, 93, 9166–9171. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Yang, K.S.; Kang, S.W.; Woo, H.A.; Chang, T.S. Controlled elimination of intracellular H2O2: Regulation of peroxiredoxin, catalase, and glutathione peroxidase via post-translational modification. Antioxid. Redox Signal. 2005, 7, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, M.A.; Robb, S.J.; Clark, J.B. Nitric oxide and Fenton/Haber-Weiss chemistry: Nitric oxide is a potent antioxidant at physiological concentrations. J. Neurochem. 2003, 87, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, B.; Pogozelski, W.K.; Tullius, T.D. DNA strand breaking by the hydroxyl radical is governed by the accessible surface areas of the hydrogen atoms of the DNA backbone. Proc. Natl. Acad. Sci. USA 1998, 95, 9738–9743. [Google Scholar] [CrossRef] [PubMed]
- Mergia, E.; Friebe, A.; Dangel, O.; Russwurm, M.; Koesling, D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J. Clin. Investig. 2006, 116, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Atochin, D.N.; Huang, P.L. Endothelial nitric oxide synthase transgenic models of endothelial dysfunction. Pflugers Arch. 2010, 460, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Atochin, D.N.; Demchenko, I.T.; Astern, J.; Boso, A.E.; Piantadosi, C.A.; Huang, P.L. Contributions of endothelial and neuronal nitric oxide synthases to cerebrovascular responses to hyperoxia. J. Cereb. Blood Flow Metab. 2003, 23, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L. Lessons learned from nitric oxide synthase knockout animals. Semin. Perinatol. 2000, 24, 87–90. [Google Scholar] [CrossRef]
- Freedman, J.E.; Sauter, R.; Battinelli, E.M.; Ault, K.; Knowles, C.; Huang, P.L.; Loscalzo, J. Deficient platelet-derived nitric oxide and enhanced hemostasis in mice lacking the NOSIII gene. Circ. Res. 1999, 84, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Kuhlencordt, P.J.; Rosel, E.; Gerszten, R.E.; Morales-Ruiz, M.; Dombkowski, D.; Atkinson, W.J.; Han, F.; Preffer, F.; Rosenzweig, A.; Sessa, W.C.; et al. Role of endothelial nitric oxide synthase in endothelial activation: Insights from eNOS knockout endothelial cells. Am. J. Physiol. Cell Physiol. 2004, 286, C1195–C1202. [Google Scholar] [CrossRef] [PubMed]
- Lefer, D.J.; Jones, S.P.; Girod, W.G.; Baines, A.; Grisham, M.B.; Cockrell, A.S.; Huang, P.L.; Scalia, R. Leukocyte-endothelial cell interactions in nitric oxide synthase-deficient mice. Am. J. Physiol. 1999, 276, H1943–H1950. [Google Scholar] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Atochin, D.N.; Wang, A.; Liu, V.W.; Critchlow, J.D.; Dantas, A.P.; Looft-Wilson, R.; Murata, T.; Salomone, S.; Shin, H.K.; Ayata, C.; et al. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J. Clin. Investig. 2007, 117, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Behnke, B.J.; Stabley, J.N.; Kilar, C.R.; Park, Y.; Narayanan, A.; Alwood, J.S.; Shirazi-Fard, Y.; Schreurs, A.S.; Globus, R.K.; et al. Effects of high-let radiation exposure and hindlimb unloading on skeletal muscle resistance artery vasomotor properties and cancellous bone microarchitecture in mice. Radiat. Res. 2016, 185, 257–276. [Google Scholar] [CrossRef] [PubMed]
- Ortega, N.; Behonick, D.J.; Werb, Z. Matrix remodeling during endochondral ossification. Trends Cell Biol. 2004, 14, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Mackie, E.J.; Ahmed, Y.A.; Tatarczuch, L.; Chen, K.S.; Mirams, M. Endochondral ossification: How cartilage is converted into bone in the developing skeleton. Int. J. Biochem. Cell Biol. 2008, 40, 46–62. [Google Scholar] [CrossRef] [PubMed]
- Kirchen, M.E.; O’Connor, K.M.; Gruber, H.E.; Sweeney, J.R.; Fras, I.A.; Stover, S.J.; Sarmiento, A.; Marshall, G.J. Effects of microgravity on bone healing in a rat fibular osteotomy model. Clin. Orthop. Relat. Res. 1995, 231–242. [Google Scholar]
- Vandamme, K.; Holy, X.; Bensidhoum, M.; Deschepper, M.; Logeart-Avramoglou, D.; Naert, I.; Duyck, J.; Petite, H. Impaired osteoblastogenesis potential of progenitor cells in skeletal unloading is associated with alterations in angiogenic and energy metabolism profile. Biomed. Mater. Eng. 2012, 22, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Fei, J.; Peyrin, F.; Malaval, L.; Vico, L.; Lafage-Proust, M.H. Imaging and quantitative assessment of long bone vascularization in the adult rat using microcomputed tomography. Anat. Rec. 2010, 293, 215–252. [Google Scholar] [CrossRef]
- Colleran, P.N.; Wilkerson, M.K.; Bloomfield, S.A.; Suva, L.J.; Turner, R.T.; Delp, M.D. Alterations in skeletal perfusion with simulated microgravity: A possible mechanism for bone remodeling. J. Appl. Physiol. 2000, 89, 1046–1054. [Google Scholar] [PubMed]
- Ramasamy, S.K.; Kusumbe, A.P.; Schiller, M.; Zeuschner, D.; Bixel, M.G.; Milia, C.; Gamrekelashvili, J.; Limbourg, A.; Medvinsky, A.; Santoro, M.M.; et al. Blood flow controls bone vascular function and osteogenesis. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Maes, C.; Carmeliet, P.; Moermans, K.; Stockmans, I.; Smets, N.; Collen, D.; Bouillon, R.; Carmeliet, G. Impaired angiogenesis and endochondral bone formation in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Mech. Dev. 2002, 111, 61–73. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, C.; Deng, L.; Liu, X.; Cao, X.; Gilbert, S.R.; Bouxsein, M.L.; Faugere, M.C.; Guldberg, R.E.; Gerstenfeld, L.C.; et al. The hypoxia-inducible factor α pathway couples angiogenesis to osteogenesis during skeletal development. J. Clin. Investig. 2007, 117, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Bungo, M.W.; Johnson, P.C., Jr. Cardiovascular examinations and observations of deconditioning during the space shuttle orbital flight test program. Aviat. Space Environ. Med. 1983, 54, 1001–1004. [Google Scholar] [PubMed]
- Mulvagh, S.L.; Charles, J.B.; Riddle, J.M.; Rehbein, T.L.; Bungo, M.W. Echocardiographic evaluation of the cardiovascular effects of short-duration spaceflight. J. Clin. Pharmacol. 1991, 31, 1024–1026. [Google Scholar] [CrossRef] [PubMed]
- Meck, J.V.; Reyes, C.J.; Perez, S.A.; Goldberger, A.L.; Ziegler, M.G. Marked exacerbation of orthostatic intolerance after long- vs. short-duration spaceflight in veteran astronauts. Psychosom. Med. 2001, 63, 865–873. [Google Scholar] [PubMed]
- Buckey, J.C., Jr.; Lane, L.D.; Levine, B.D.; Watenpaugh, D.E.; Wright, S.J.; Moore, W.E.; Gaffney, F.A.; Blomqvist, C.G. Orthostatic intolerance after spaceflight. J. Appl. Physiol. 1996, 81, 7–18. [Google Scholar] [PubMed]
- Lee, S.M.; Feiveson, A.H.; Stein, S.; Stenger, M.B.; Platts, S.H. Orthostatic intolerance after ISS and space shuttle missions. Aerosp. Med. Hum. Perform. 2015, 86 (Suppl. S12), 54–67. [Google Scholar] [CrossRef] [PubMed]
- Stabley, J.N.; Dominguez, J.M., 2nd; Dominguez, C.E.; Mora Solis, F.R.; Ahlgren, J.; Behnke, B.J.; Muller-Delp, J.M.; Delp, M.D. Spaceflight reduces vasoconstrictor responsiveness of skeletal muscle resistance arteries in mice. J. Appl. Physiol. 2012, 113, 1439–1445. [Google Scholar] [CrossRef] [PubMed]
- Delp, M.D. Myogenic and vasoconstrictor responsiveness of skeletal muscle arterioles is diminished by hindlimb unloading. J. Appl. Physiol. 1999, 86, 1178–1184. [Google Scholar] [PubMed]
- Prisby, R.D.; Alwood, J.S.; Behnke, B.J.; Stabley, J.N.; McCullough, D.J.; Ghosh, P.; Globus, R.K.; Delp, M.D. Effects of hindlimb unloading and ionizing radiation on skeletal muscle resistance artery vasodilation and its relation to cancellous bone in mice. J. Appl. Physiol. 2016, 120, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Jasperse, J.L.; Woodman, C.R.; Price, E.M.; Hasser, E.M.; Laughlin, M.H. Hindlimb unweighting decreases ecNOS gene expression and endothelium-dependent dilation in rat soleus feed arteries. J. Appl. Physiol. 1999, 87, 1476–1482. [Google Scholar] [PubMed]
- Woodman, C.R.; Schrage, W.G.; Rush, J.W.; Ray, C.A.; Price, E.M.; Hasser, E.M.; Laughlin, M.H. Hindlimb unweighting decreases endothelium-dependent dilation and eNOS expression in soleus not gastrocnemius. J. Appl. Physiol. 2001, 91, 1091–1098. [Google Scholar] [PubMed]
- Prisby, R.D.; Behnke, B.J.; Allen, M.R.; Delp, M.D. Effects of skeletal unloading on the vasomotor properties of the rat femur principal nutrient artery. J. Appl. Physiol. 2015, 118, 980–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplanskii, A.S.; Durnova, G.N.; Sakharova, Z.F.; Il’ina-Kakueva, E.I. [Histomorphometric analysis of the bones of rats on board the Kosmos 1667 biosatellite]. Kosm. Biol. Aviakosm. Med. 1987, 21, 25–31. [Google Scholar] [PubMed]
- Lueken, S.A.; Arnaud, S.B.; Taylor, A.K.; Baylink, D.J. Changes in markers of bone formation and resorption in a bed rest model of weightlessness. J. Bone Miner. Res. 1993, 8, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Shahnazari, M.; Kurimoto, P.; Boudignon, B.M.; Orwoll, B.E.; Bikle, D.D.; Halloran, B.P. Simulated spaceflight produces a rapid and sustained loss of osteoprogenitors and an acute but transitory rise of osteoclast precursors in two genetic strains of mice. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1354–E1362. [Google Scholar] [CrossRef] [PubMed]
- Delp, M.D.; Charvat, J.M.; Limoli, C.L.; Globus, R.K.; Ghosh, P. apollo lunar astronauts show higher cardiovascular disease mortality: Possible deep space radiation effects on the vascular endothelium. Sci Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev. Cell 2008, 14, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Damulewicz, M.; Pyza, E.; Jozkowicz, A.; Dulak, J. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: An evolutionarily conserved mechanism. Cell. Mol. Life Sci. 2016, 73, 3221–3247. [Google Scholar] [CrossRef] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the β-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Davies, K.J.A.; Forman, H.J. Oxidative stress response and Nrf2 signaling in aging. Free Radic. Biol. Med. 2015, 88, 314–336. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Lu, R.; Chang, J.C.; Kan, Y.W. NRF2, a member of the NFE2 family of transcription factors, is not essential for murine erythropoiesis, growth, and development. Proc. Natl. Acad. Sci. USA 1996, 93, 13943–13948. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, G.G.; Cregor, M.; McAndrews, K.; Morales, C.C.; McCabe, L.D.; McCabe, G.P.; Peacock, M.; Burr, D.; Weaver, C.; Bellido, T. Nrf2 regulates mass accrual and the antioxidant endogenous response in bone differently depending on the sex and age. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Kim, K.; Norris, A.J.; Vlashi, E.; Phillips, T.M.; Lagadec, C.; della Donna, L.; Ratikan, J.; Szelag, H.; Hlatky, L.; et al. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010, 70, 8886–8895. [Google Scholar] [CrossRef] [PubMed]
- Strom, J.; Chen, Q.M. Loss of Nrf2 promotes rapid progression to heart failure following myocardial infarction. Toxicol. Appl. Pharmacol. 2017, 327, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Larabee, C.M.; Desai, S.; Agasing, A.; Georgescu, C.; Wren, J.D.; Axtell, R.C.; Plafker, S.M. Loss of Nrf2 exacerbates the visual deficits and optic neuritis elicited by experimental autoimmune encephalomyelitis. Mol. Vis. 2016, 22, 1503–1513. [Google Scholar] [PubMed]
- Zhao, Z.; Chen, Y.; Wang, J.; Sternberg, P.; Freeman, M.L.; Grossniklaus, H.E.; Cai, J. Age-related retinopathy in NRF2-deficient mice. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Singhal, V.; Biswal, S.; Thimmulappa, R.K.; DiGirolamo, D.J. Nrf2 is required for normal postnatal bone acquisition in mice. Bone Res. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Rana, T.; Schultz, M.A.; Freeman, M.L.; Biswas, S. Loss of Nrf2 accelerates ionizing radiation-induced bone loss by upregulating RANKL. Free Radic. Biol Med. 2012, 53, 2298–2307. [Google Scholar] [CrossRef] [PubMed]
- Gremmels, H.; de Jong, O.G.; Hazenbrink, D.H.; Fledderus, J.O.; Verhaar, M.C. The transcription factor Nrf2 protects angiogenic capacity of endothelial colony-forming cells in high-oxygen radical stress conditions. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, S.; Lee, H.; Yang, Y.; Jeong, W. Nrf2 deficiency induces oxidative stress and promotes RANKL-induced osteoclast differentiation. Free Radic. Biol. Med. 2013, 65, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Park, C.K.; Lee, Y.; Kim, K.H.; Lee, Z.H.; Joo, M.; Kim, H.H. Nrf2 is a novel regulator of bone acquisition. Bone 2014, 63, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.X.; Li, L.; Corry, K.A.; Zhang, P.; Yang, Y.; Himes, E.; Mihuti, C.L.; Nelson, C.; Dai, G.; Li, J. Deletion of Nrf2 reduces skeletal mechanical properties and decreases load-driven bone formation. Bone 2015, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Leach, C.S. A review of the consequences of fluid and electrolyte shifts in weightlessness. Acta Astronaut. 1979, 6, 1123–1135. [Google Scholar] [CrossRef]
- Natochin, Y.V.; Kozyrevskaya, G.I.; Grigor’yev, A.I. Study of water-salt metabolism and renal function in cosmonauts. Acta Astronaut. 1975, 2, 175–188. [Google Scholar] [CrossRef]
- Grigor’ev, A.I.; Dorokhova, B.P.; Semenov, V.; Morukov, B.V.; Baichorov, E.O. [Water-electrolyte balance and functional state of the kidneys in cosmonauts after a 185-day space flight]. Kosm. Biol. Aviakosm. Med. 1985, 19, 17–21. [Google Scholar]
- Grigoriev, A.I.; Morukov, B.V.; Vorobiev, D.V. Water and electrolyte studies during long-term missions onboard the space stations SALYUT and MIR. Clin. Investig. 1994, 72, 169–198. [Google Scholar] [CrossRef] [PubMed]
- Priestley, J.R.; Kautenburg, K.E.; Casati, M.C.; Endres, B.T.; Geurts, A.M.; Lombard, J.H. The NRF2 knockout rat: A new animal model to study endothelial dysfunction, oxidant stress, and microvascular rarefaction. Am. J. Physiol. Heart Circ. Physiol. 2016, 310, H478–H488. [Google Scholar] [CrossRef] [PubMed]
- Smietana, M.J.; Arruda, E.M.; Faulkner, J.A.; Brooks, S.V.; Larkin, L.M. Reactive oxygen species on bone mineral density and mechanics in Cu,Zn superoxide dismutase (Sod1) knockout mice. Biochem. Biophys. Res. Commun. 2010, 403, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., 2nd; Csete, M.; et al. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Schriner, S.E.; Linford, N.J.; Martin, G.M.; Treuting, P.; Ogburn, C.E.; Emond, M.; Coskun, P.E.; Ladiges, W.; Wolf, N.; van Remmen, H.; et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005, 308, 1909–1911. [Google Scholar] [CrossRef] [PubMed]
- Treuting, P.M.; Linford, N.J.; Knoblaugh, S.E.; Emond, M.J.; Morton, J.F.; Martin, G.M.; Rabinovitch, P.S.; Ladiges, W.C. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2008, 63, 813–832. [Google Scholar] [CrossRef] [PubMed]
- Mao, P.; Manczak, M.; Calkins, M.J.; Truong, Q.; Reddy, T.P.; Reddy, A.P.; Shirendeb, U.; Lo, H.H.; Rabinovitch, P.S.; Reddy, P.H. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Hum. Mol. Genet. 2012, 21, 2973–2990. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Wanagat, J.; Laflamme, M.; Marcinek, D.J.; Emond, M.J.; Ngo, C.P.; Prolla, T.A.; Rabinovitch, P.S. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 2010, 9, 536–564. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy 2011, 7, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, G.Z.; Rabinovitch, P.S.; Tabas, I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nuclear factor-κB-mediated inflammation in macrophages. Circ. Res. 2014, 114, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Ucer, S.; Iyer, S.; Kim, H.N.; Han, L.; Rutlen, C.; Allison, K.; Thostenson, J.D.; de Cabo, R.; Jilka, R.L.; O’Brien, C.; et al. The effects of aging and sex steroid deficiency on the murine skeleton are independent and mechanistically distinct. J. Bone Miner. Res. 2017, 32, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Liao, A.C.; Craver, B.M.; Tseng, B.P.; Tran, K.K.; Parihar, V.K.; Acharya, M.M.; Limoli, C.L. Mitochondrial-targeted human catalase affords neuroprotection from proton irradiation. Radiat. Res. 2013, 180, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Parihar, V.K.; Allen, B.D.; Tran, K.K.; Chmielewski, N.N.; Craver, B.M.; Martirosian, V.; Morganti, J.M.; Rosi, S.; Vlkolinsky, R.; Acharya, M.M.; et al. Targeted overexpression of mitochondrial catalase prevents radiation-induced cognitive dysfunction. Antioxid. Redox Signal. 2015, 22, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, N.N.; Caressi, C.; Giedzinski, E.; Parihar, V.K.; Limoli, C.L. Contrasting the effects of proton irradiation on dendritic complexity of subiculum neurons in wild type and MCAT mice. Environ. Mol. Mutagen. 2016, 57, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Feng, W.; Wang, Y.; Luo, Y.; Allen, A.R.; Koturbash, I.; Turner, J.; Stewart, B.; Raber, J.; Hauer-Jensen, M.; et al. Whole-body proton irradiation causes long-term damage to hematopoietic stem cells in mice. Radiat. Res. 2015, 183, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ran, Q.; Xiang, Y.; Xiang, L.; Chen, L.; Li, F.; Wu, J.; Wu, C.; Li, Z. Co-activation of PKC-Δ by CRIF1 modulates oxidative stress in bone marrow multipotent mesenchymal stromal cells after Irradiation by phosphorylating NRF2 ser40. Theranostics 2017, 7, 2634–2648. [Google Scholar] [CrossRef] [PubMed]
- Alwood, J.S.; Yumoto, K.; Mojarrab, R.; Limoli, C.L.; Almeida, E.A.; Searby, N.D.; Globus, R.K. Heavy ion irradiation and unloading effects on mouse lumbar vertebral microarchitecture, mechanical properties and tissue stresses. Bone 2010, 47, 248–285. [Google Scholar] [CrossRef] [PubMed]
- Yumoto, K.; Globus, R.K.; Mojarrab, R.; Arakaki, J.; Wang, A.; Searby, N.D.; Almeida, E.A.; Limoli, C.L. Short-term effects of whole-body exposure to (56)fe ions in combination with musculoskeletal disuse on bone cells. Radiat. Res. 2010, 173, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, S.A.; Bandstra, E.R.; Willey, J.S.; Riffle, S.E.; Tirado-Lee, L.; Nelson, G.A.; Pecaut, M.J.; Bateman, T.A. Effect of proton irradiation followed by hindlimb unloading on bone in mature mice: A model of long-duration spaceflight. Bone 2012, 51, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Alwood, J.S.; Kumar, A.; Tran, L.H.; Wang, A.; Limoli, C.L.; Globus, R.K. Low-dose, ionizing radiation and age-related changes in skeletal microarchitecture. J. Aging Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Stewart, J.; Ware, J.H.; Zhou, Z.; Donahue, J.J.; Kennedy, A.R. Effects of dietary supplements on the space radiation-induced reduction in total antioxidant status in CBA mice. Radiat. Res. 2006, 165, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.R.; Davis, J.G.; Carlton, W.; Ware, J.H. Effects of dietary antioxidant supplementation on the development of malignant lymphoma and other neoplastic lesions in mice exposed to proton or iron-ion radiation. Radiat. Res. 2008, 169, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Wambi, C.; Sanzari, J.; Wan, X.S.; Nuth, M.; Davis, J.; Ko, Y.H.; Sayers, C.M.; Baran, M.; Ware, J.H.; Kennedy, A.R. Dietary antioxidants protect hematopoietic cells and improve animal survival after total-body irradiation. Radiat. Res. 2008, 169, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Matsugo, S.; Yan, L.J.; Han, D.; Trischler, H.J.; Packer, L. Elucidation of antioxidant activity of α lipoic acid toward hydroxyl radical. Biochem. Biophys. Res. Commun. 1995, 208, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.L.; Kolozsvary, A.; Liu, J.; Jenrow, K.A.; Ryu, S.; Kim, J.H. Antioxidant diet supplementation starting 24 hours after exposure reduces radiation lethality. Radiat. Res. 2010, 173, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Espirito Santo, A.I.; Ersek, A.; Freidin, A.; Feldmann, M.; Stoop, A.A.; Horwood, N.J. Selective inhibition of TNFR1 reduces osteoclast numbers and is differentiated from anti-TNF in a LPS-driven model of inflammatory bone loss. Biochem. Biophys. Res. Commun. 2015, 464, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R.; Purdue, P.E.; Crotti, T.N.; Shen, Z.; Flannery, M.R.; Binder, N.B.; Ross, F.P.; McHugh, K.P. Bone remodelling in inflammatory arthritis. Ann. Rheum. Dis. 2013, 72 (Suppl. S2). [Google Scholar] [CrossRef] [PubMed]
- Vinson, J.A.; Zubik, L.; Bose, P.; Samman, N.; Proch, J. Dried fruits: Excellent in vitro and in vivo antioxidants. J. Am. Coll. Nutr. 2005, 24, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Halloran, B.P.; Wronski, T.J.; VonHerzen, D.C.; Chu, V.; Xia, X.; Pingel, J.E.; Williams, A.A.; Smith, B.J. Dietary dried plum increases bone mass in adult and aged male mice. J. Nutr. 2010, 140, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Lucas, E.A.; Hooshmand, S.; Campbell, S.; Akhter, M.P.; Arjmandi, B.H. Addition of fructooligosaccharides and dried plum to soy-based diets reverses bone loss in the ovariectomized rat. Evid. Based Complement. Alternat. Med. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Rendina, E.; Lim, Y.F.; Marlow, D.; Wang, Y.; Clarke, S.L.; Kuvibidila, S.; Lucas, E.A.; Smith, B.J. Dietary supplementation with dried plum prevents ovariectomy-induced bone loss while modulating the immune response in C57BL/6J mice. J. Nutr. Biochem. 2012, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Okumura, H.; Guo, R.; Naruse, K. Effect of oxidative stress on cardiovascular system in response to gravity. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahimic, C.G.T.; Globus, R.K. Redox Signaling and Its Impact on Skeletal and Vascular Responses to Spaceflight. Int. J. Mol. Sci. 2017, 18, 2153. https://doi.org/10.3390/ijms18102153
Tahimic CGT, Globus RK. Redox Signaling and Its Impact on Skeletal and Vascular Responses to Spaceflight. International Journal of Molecular Sciences. 2017; 18(10):2153. https://doi.org/10.3390/ijms18102153
Chicago/Turabian StyleTahimic, Candice G. T., and Ruth K. Globus. 2017. "Redox Signaling and Its Impact on Skeletal and Vascular Responses to Spaceflight" International Journal of Molecular Sciences 18, no. 10: 2153. https://doi.org/10.3390/ijms18102153
APA StyleTahimic, C. G. T., & Globus, R. K. (2017). Redox Signaling and Its Impact on Skeletal and Vascular Responses to Spaceflight. International Journal of Molecular Sciences, 18(10), 2153. https://doi.org/10.3390/ijms18102153