Reduced Abundance and Subverted Functions of Proteins in Prion-Like Diseases: Gained Functions Fascinate but Lost Functions Affect Aetiology


Abstract
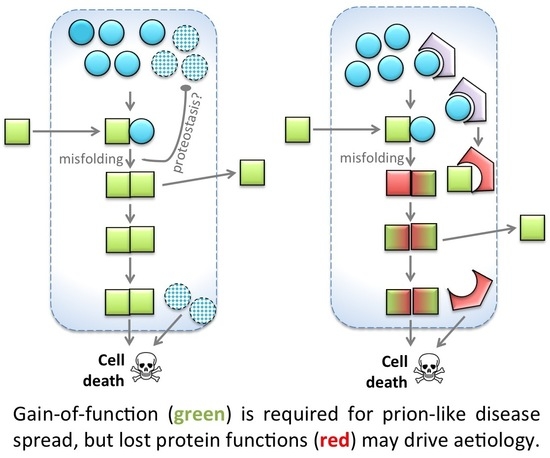
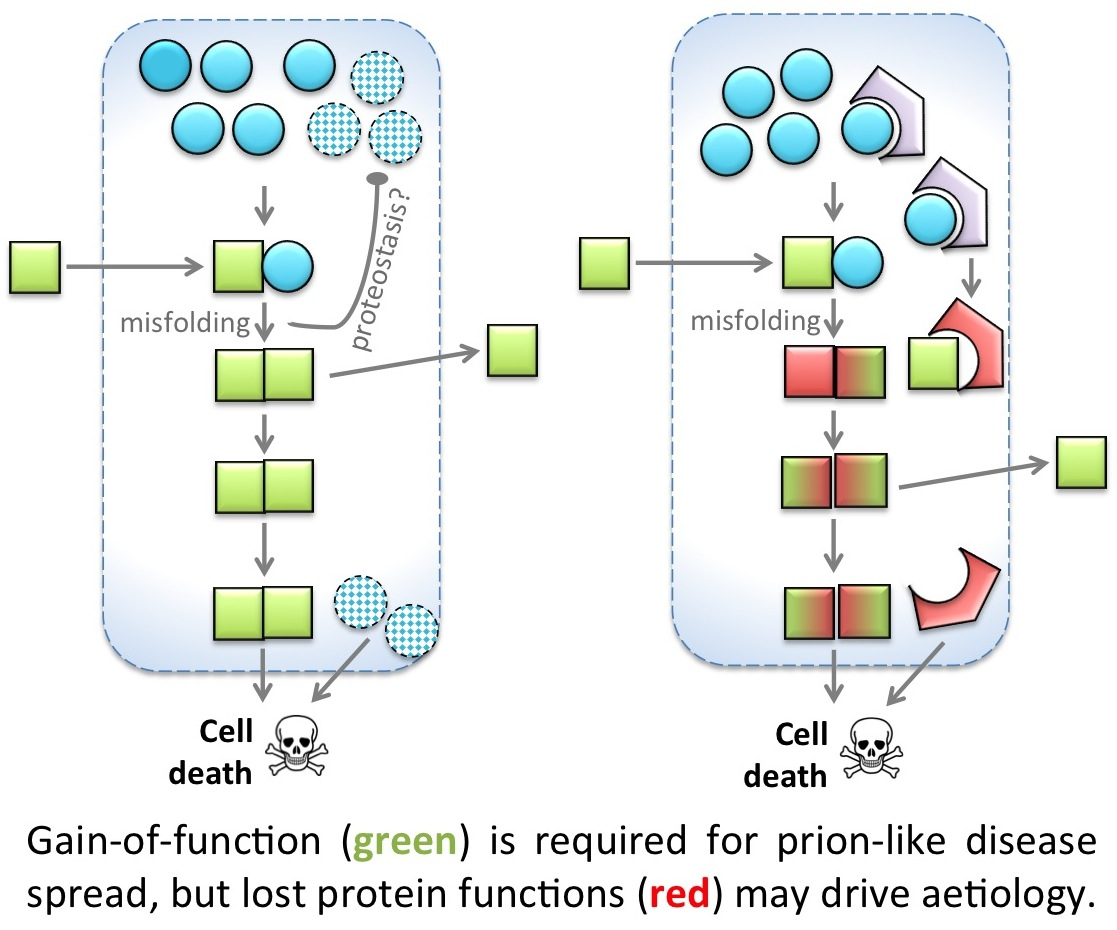
:
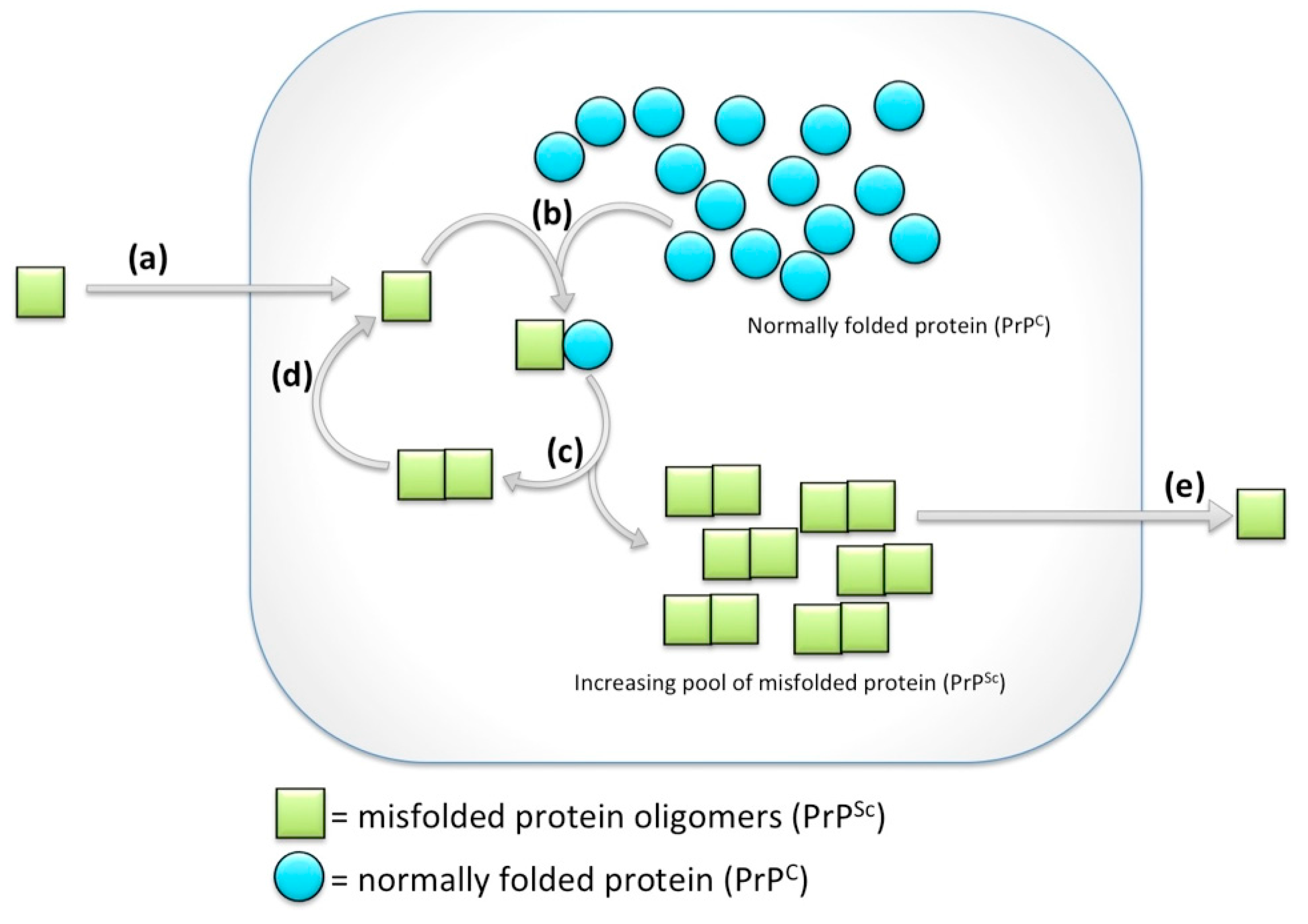
{kind=link}
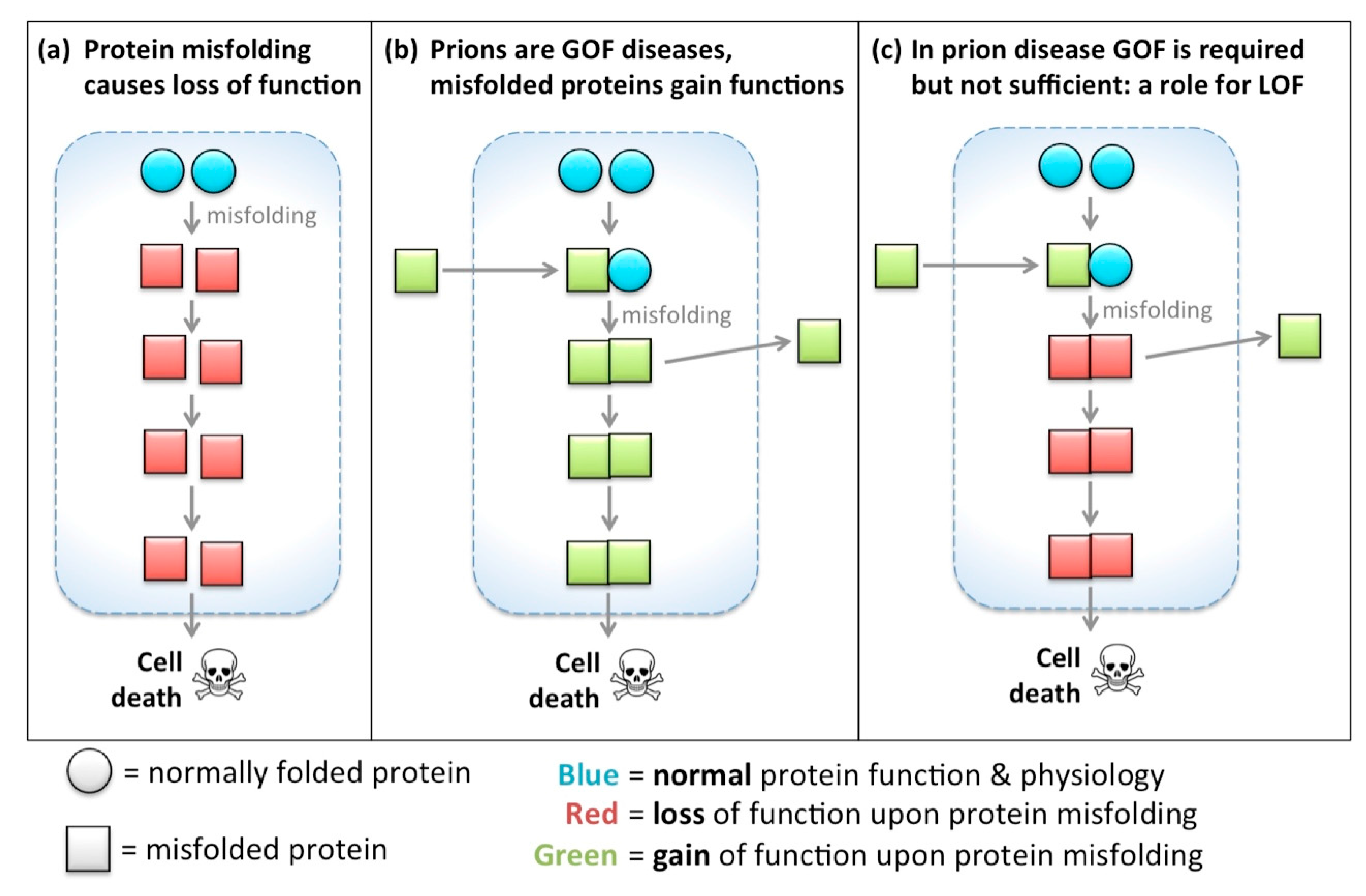
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Prion-Like Diseases
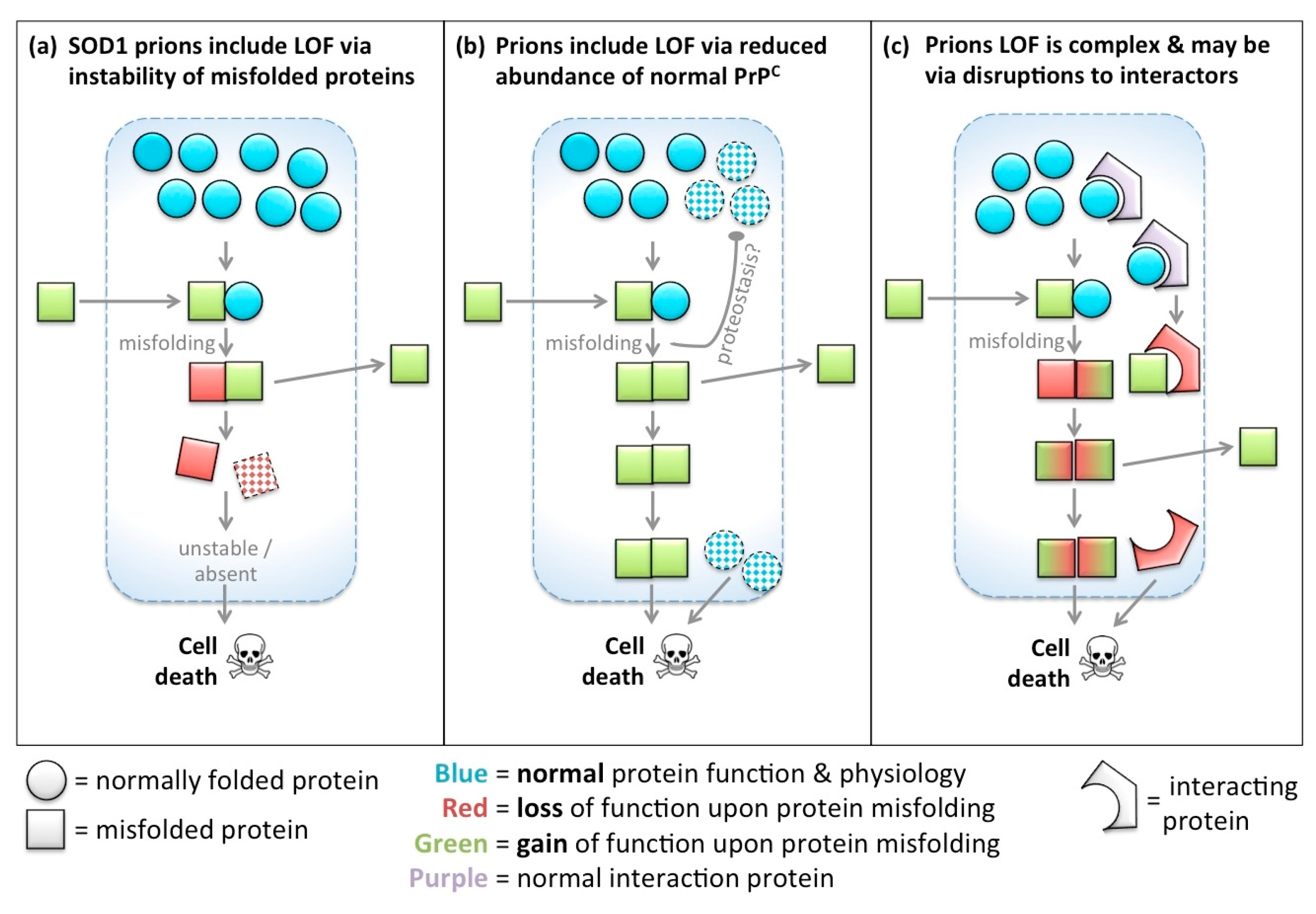
3. Protein Functions are Lost, Reduced and Subverted upon Protein Misfolding: Yet Gained Functions Fascinate the Prion Biologist
- H0: Gain of function is required in prion disease, but not sufficient.
- H0: Loss of function is not sufficient in prion disease, but is required for progression.
4. Physiological Roles for PrPC: Lost Functions Resemble Disease Etiology
5. Physiological Roles for SOD1: Functions Whose Loss Can Rationally Explain Etiology
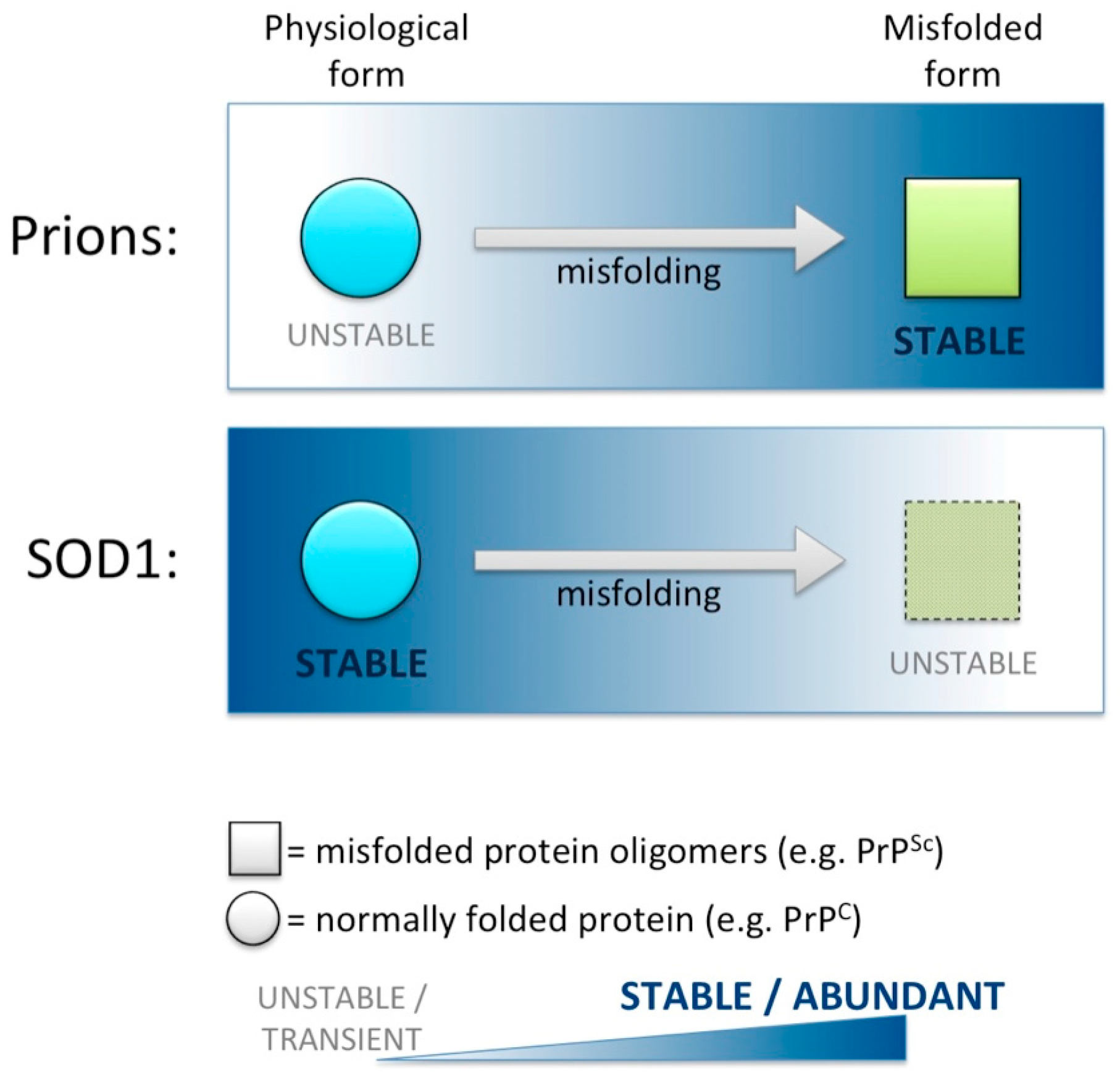
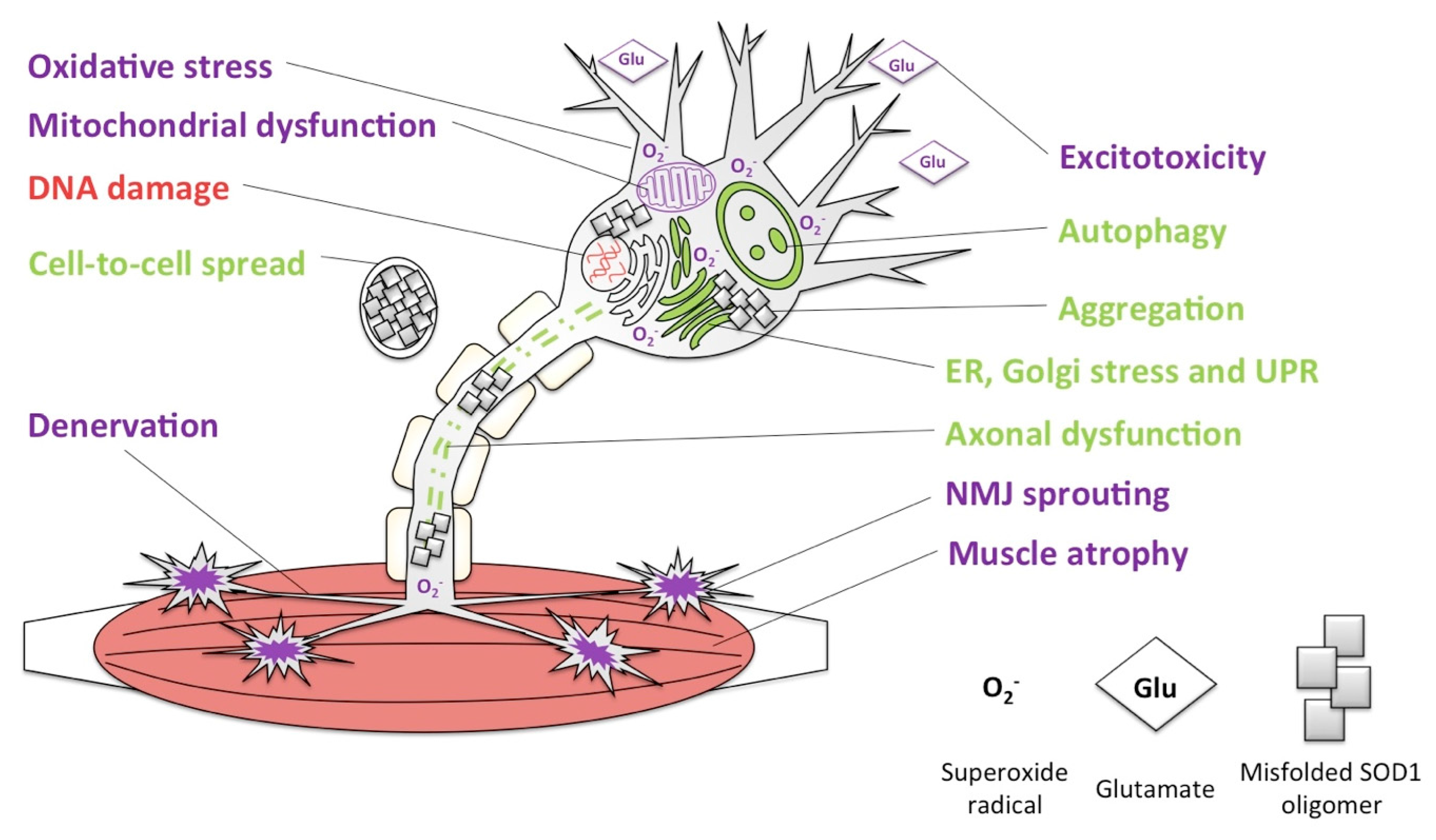
6. Loss of SOD1 Following Misfolding: Similarities in Prion but Not Other Prion-Like Diseases?
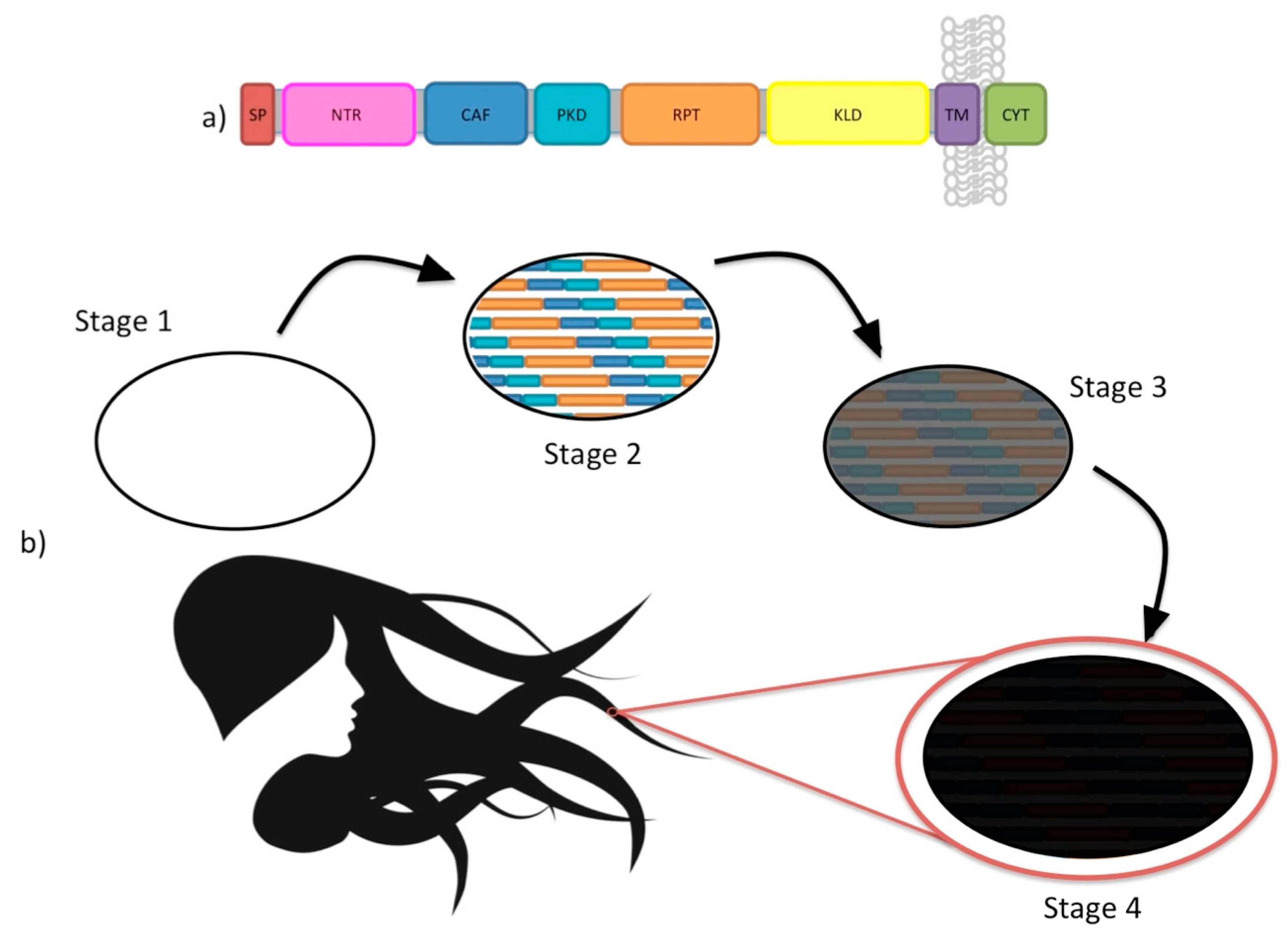
7. Loss and Gain of Function in Amyloidosis: What News from Functional Amyloid?
8. Challenges of Disentangling GOF from LOF: Experiments Cannot Prove that GOF Is Sufficient
9. Interventions to Test If Loss of Function Plays a Role in Prion-Like Diseases
10. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer Disease |
| ALS | Amyotrophic Lateral Sclerosis |
| Aβ | Amyloid-β |
| AβPP | Amyloid-β Precursor Protein |
| CWD | Chronic Wasting Disease |
| GOF | Gain of Function (following protein misfolding/aggregation) |
| LOF | Loss of Function (following protein misfolding/aggregation) |
| PrPC | Prion Protein, cellular form (normally folded form in healthy brains) |
| PrPSc | Prion Protein, Scrapie form (misfolded form, disease-causing) |
| SOD1 | Superoxide Dismutase 1 |
| TDM | Template Directed Misfolding |
References
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Falcon, B.; Clavaguera, F.; Tolnay, M. Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr. Neurol. Neurosci. Rep. 2014, 14, 495. [Google Scholar] [CrossRef] [PubMed]
- Hall, G.F.; Patuto, B.A. Is tau ready for admission to the prion club? Prion 2012, 6, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Marciniuk, K.; Taschuk, R.; Napper, S. Evidence for prion-like mechanisms in several neurodegenerative diseases: Potential implications for immunotherapy. Clin. Dev. Immunol. 2013, 2013, 473706. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.L.; Allison, W.T. Protein misfolding in prion and prion-like diseases: Reconsidering a required role for protein loss-of-function. J. Alzheimers Dis. 2016, 54, 3–29. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Neurodegeneration. Alzheimer’s and parkinson’s diseases: The prion concept in relation to assembled abeta, tau, and alpha-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Abeliovich, A.; Gitler, A.D. Defects in trafficking bridge parkinson’s disease pathology and genetics. Nature 2016, 539, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to cftr processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Yang, X.; Zhao, Y.; Petersen, R.B.; Liu, X.; Liu, Y.; Huang, K. Amyloidogenicity of p53: A hidden link between protein misfolding and cancer. Curr. Protein Pept. Sci. 2015, 16, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Rangel, L.P.; Costa, D.C.; Cordeiro, Y.; De Moura Gallo, C.V. Expanding the prion concept to cancer biology: Dominant-negative effect of aggregates of mutant p53 tumour suppressor. Biosci. Rep. 2013, 33, e00054. [Google Scholar] [CrossRef] [PubMed]
- Ano Bom, A.P.; Rangel, L.P.; Costa, D.C.; de Oliveira, G.A.; Sanches, D.; Braga, C.A.; Gava, L.M.; Ramos, C.H.; Cepeda, A.O.; Stumbo, A.C.; et al. Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: Implications for cancer. J. Biol. Chem. 2012, 287, 28152–28162. [Google Scholar] [CrossRef] [PubMed]
- Rangel, L.P.; Costa, D.C.; Vieira, T.C.; Silva, J.L. The aggregation of mutant p53 produces prion-like properties in cancer. Prion 2014, 8, 75–84. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.A.; Rangel, L.P.; Costa, D.C.; Silva, J.L. Misfolding, aggregation, and disordered segments in c-abl and p53 in human cancer. Front. Oncol. 2015, 5, 97. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Murshid, A. Molecular chaperone accumulation in cancer and decrease in Alzheimer’s disease: The potential roles of hsf1. Front. Neurosci. 2017, 11, 192. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Cohen-Rosenzweig, C.; Ciechanover, A. The ubiquitin-proteasome system and autophagy: Coordinated and independent activities. Int. J. Biochem. Cell. Biol. 2016, 79, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Morales-Scheihing, D.; Butler, P.C.; Soto, C. Type 2 diabetes as a protein misfolding disease. Trends Mol. Med. 2015, 21, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.A.; de Oliveira, G.A. Cardiac troponin and tropomyosin: Structural and cellular perspectives to unveil the hypertrophic cardiomyopathy phenotype. Front. Physiol. 2016, 7, 429. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, K.R.; McMillan, J.D.; Kumar, A. Emerging roles of er stress and unfolded protein response pathways in skeletal muscle health and disease. J. Cell. Physiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Vanden Broeck, L.; Callaerts, P.; Dermaut, B. Tdp-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Saborio, G.P.; Anderes, L. Cyclic amplification of protein misfolding: Application to prion-related disorders and beyond. Trends Neurosci. 2002, 25, 390–394. [Google Scholar] [CrossRef]
- Zanusso, G.; Monaco, S.; Pocchiari, M.; Caughey, B. Advanced tests for early and accurate diagnosis of creutzfeldt-jakob disease. Nat. Rev. Neurol. 2016, 12, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Orru, C.D.; Wilham, J.M.; Vascellari, S.; Hughson, A.G.; Caughey, B. New generation quic assays for prion seeding activity. Prion 2012, 6, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Salvadores, N.; Shahnawaz, M.; Scarpini, E.; Tagliavini, F.; Soto, C. Detection of misfolded abeta oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell. Rep. 2014, 7, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Shahnawaz, M.; Tokuda, T.; Waragai, M.; Mendez, N.; Ishii, R.; Trenkwalder, C.; Mollenhauer, B.; Soto, C. Development of a biochemical diagnosis of parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017, 74, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Herva, M.E.; Zibaee, S.; Fraser, G.; Barker, R.A.; Goedert, M.; Spillantini, M.G. Anti-amyloid compounds inhibit alpha-synuclein aggregation induced by protein misfolding cyclic amplification (pmca). J. Biol. Chem. 2014, 289, 11897–11905. [Google Scholar] [CrossRef] [PubMed]
- Fairfoul, G.; McGuire, L.I.; Pal, S.; Ironside, J.W.; Neumann, J.; Christie, S.; Joachim, C.; Esiri, M.; Evetts, S.G.; Rolinski, M.; et al. Alpha-synuclein rt-quic in the csf of patients with alpha-synucleinopathies. Ann. Clin. Transl. Neurol. 2016, 3, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Canter, R.G.; Penney, J.; Tsai, L.H. The road to restoring neural circuits for the treatment of Alzheimer’s disease. Nature 2016, 539, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Bennett, D.A.; Schneider, J.A.; Arvanitakis, Z.; Wilson, R.S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 2012, 9, 628–645. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Medicine. Prion strain mutation and selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C. Proteopathic strains and the heterogeneity of neurodegenerative diseases. Annu. Rev. Genet. 2016, 50, 329–346. [Google Scholar] [CrossRef] [PubMed]
- Morange, M. The protein side of the central dogma: Permanence and change. Hist. Philos. Life Sci. 2006, 28, 513–524. [Google Scholar] [PubMed]
- Koonin, E.V. Does the central dogma still stand? Biol. Direct. 2012, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L. Disease transmission by misfolded prion-protein isoforms, prion-like amyloids, functional amyloids and the central dogma. Biology (Basel) 2016, 5, 2. [Google Scholar] [CrossRef] [PubMed]
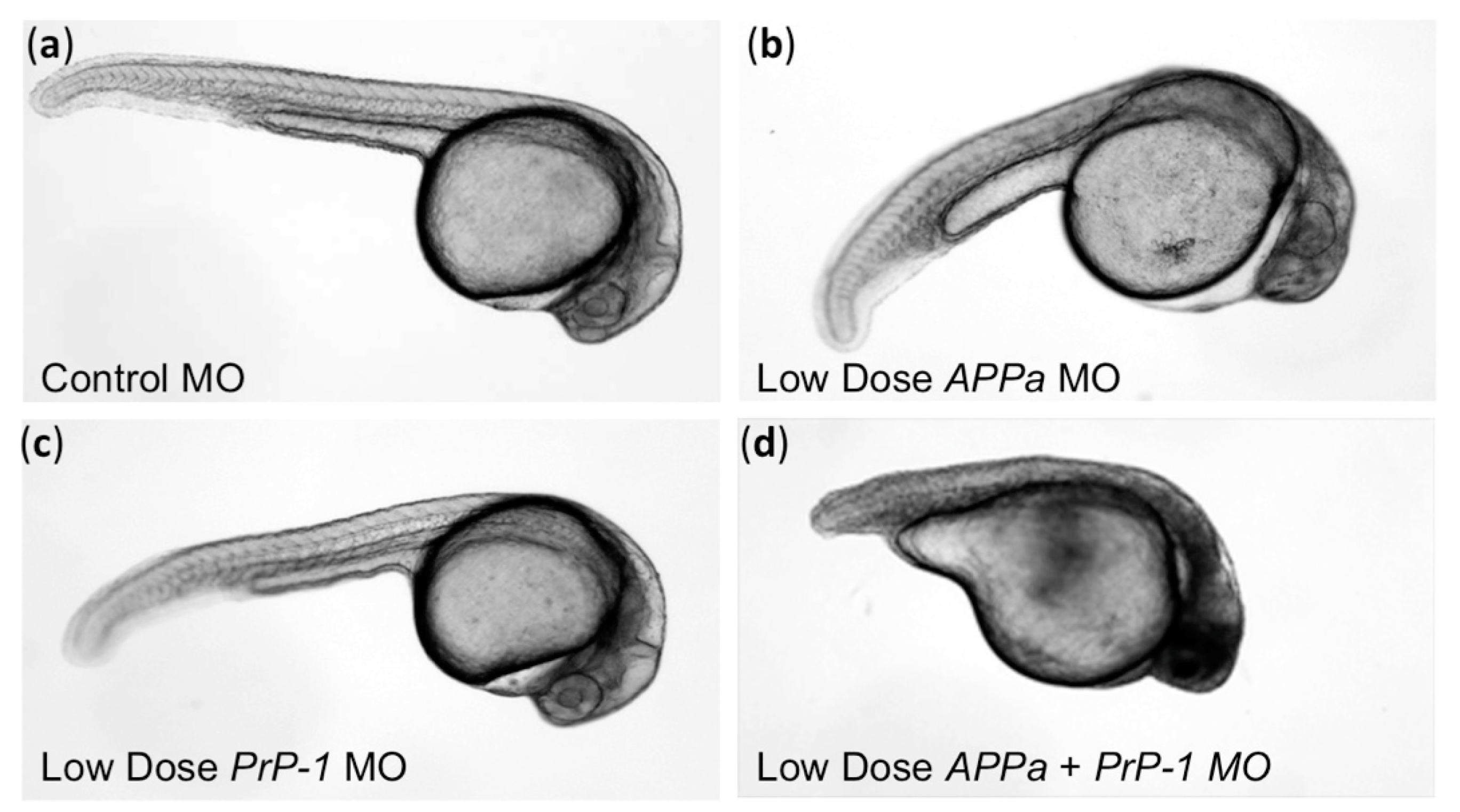
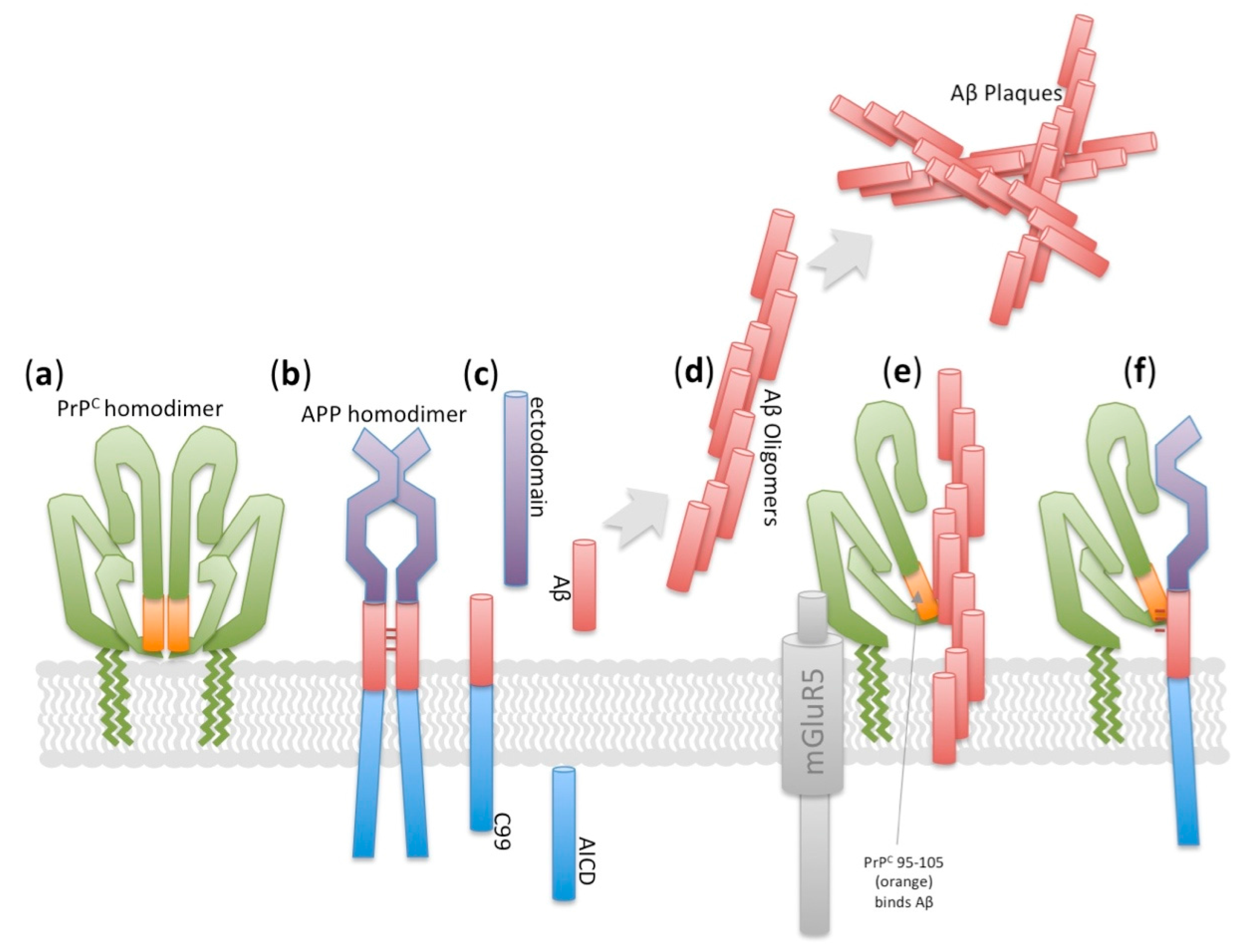
- Kaiser, D.M.; Acharya, M.; Leighton, P.L.; Wang, H.; Daude, N.; Wohlgemuth, S.; Shi, B.; Allison, W.T. Amyloid beta precursor protein and prion protein have a conserved interaction affecting cell adhesion and cns development. PLoS ONE 2012, 7, e51305. [Google Scholar] [CrossRef] [PubMed]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Stahl, N.; Borchelt, D.R.; Hsiao, K.; Prusiner, S.B. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 1987, 51, 229–240. [Google Scholar] [CrossRef]
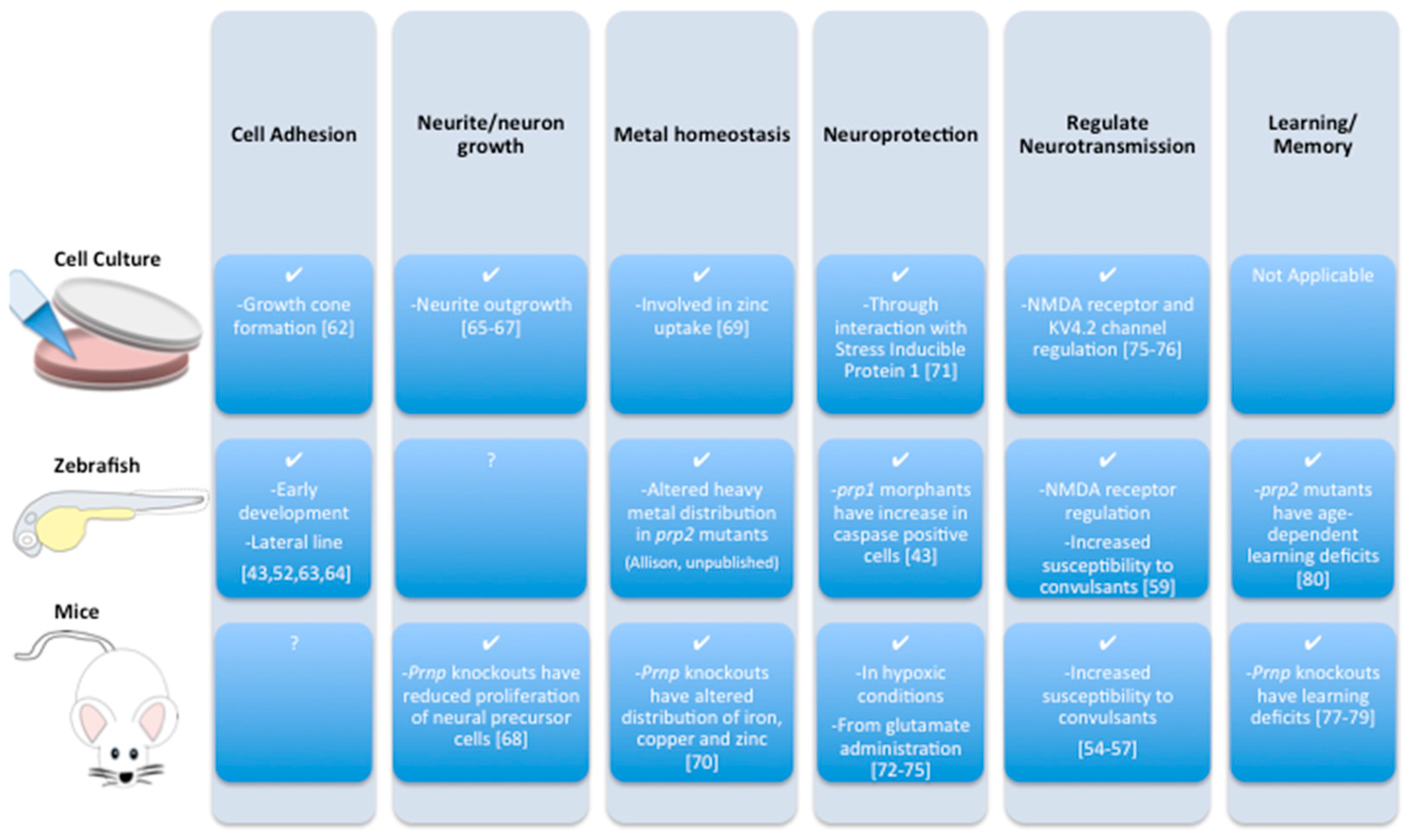
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Kanata, E.; Ouzounis, C.A.; Gilch, S.; Schatzl, H.; Sklaviadis, T. Assessing proteinase k resistance of fish prion proteins in a scrapie-infected mouse neuroblastoma cell line. Viruses 2014, 6, 4398–4421. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu. Rev. Neurosci. 2008, 31, 439–477. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; Dearmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behavior of mice lacking the neuronal cell-surface Prp protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.C.; Clarke, A.R.; Hooper, M.L.; Aitchison, L.; McConnell, I.; Hope, J. 129/ola mice carrying a null mutation in Prp that abolishes mrna production are developmentally normal. Mol. Neurobiol. 1994, 8, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Lindquist, S.; Aguzzi, A. The prion protein knockout mouse: A phenotype under challenge. Prion 2007, 1, 83–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaga-Trillo, E.; Solis, G.P.; Schrock, Y.; Geiss, C.; Luncz, L.; Thomanetz, V.; Stuermer, C.A.O. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009, 7, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Wieser, H.G.; Schindler, K.; Zumsteg, D. Eeg in creutzfeldt-jakob disease. Clin. Neurophysiol. 2006, 117, 935–951. [Google Scholar] [CrossRef] [PubMed]
- Carulla, P.; Bribian, A.; Rangel, A.; Gavin, R.; Ferrer, I.; Caelles, C.; Del Rio, J.A.; Llorens, F. Neuroprotective role of prpc against kainate-induced epileptic seizures and cell death depends on the modulation of JNK3 activation by glur6/7-PSD-95 binding. Mol. Biol. Cell 2011, 22, 3041–3054. [Google Scholar] [CrossRef] [PubMed]
- Carulla, P.; Llorens, F.; Matamoros-Angles, A.; Aguilar-Calvo, P.; Espinosa, J.C.; Gavin, R.; Ferrer, I.; Legname, G.; Torres, J.M.; del Rio, J.A. Involvement of Prp(c) in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 2015, 5, 11971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel, A.; Burgaya, F.; Gavin, R.; Soriano, E.; Aguzzi, A.; Del Rio, J.A. Enhanced susceptibility of prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of ampa/kainate receptors. J. Neurosci. Res. 2007, 85, 2741–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, R.; Amaral, O.B.; Rockenbach, I.C.; Roesler, R.; Izquierdo, I.; Cavalheiro, E.A.; Martins, V.R.; Brentani, R.R. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 1999, 40, 1679–1682. [Google Scholar] [CrossRef] [PubMed]
- Walz, R.; Castro, R.M.; Velasco, T.R.; Carlotti, C.G., Jr.; Sakamoto, A.C.; Brentani, R.R.; Martins, V.R. Cellular prion protein: Implications in seizures and epilepsy. Cell. Mol. Neurobiol. 2002, 22, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Fleisch, V.C.; Leighton, P.L.; Wang, H.; Pillay, L.M.; Ritzel, R.G.; Bhinder, G.; Roy, B.; Tierney, K.B.; Ali, D.W.; Waskiewicz, A.J.; et al. Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 2013, 55, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Caine, D.; Tinelli, R.J.; Hyare, H.; De Vita, E.; Lowe, J.; Lukic, A.; Thompson, A.; Porter, M.C.; Cipolotti, L.; Rudge, P.; et al. The cognitive profile of prion disease: A prospective clinical and imaging study. Ann. Clin. Transl. Neurol. 2015, 2, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Bodrikov, V.; Solis, G.P.; Stuermer, C.A. Prion protein promotes growth cone development through reggie/flotillin-dependent n-cadherin trafficking. J. Neurosci. 2011, 31, 18013–18025. [Google Scholar] [CrossRef] [PubMed]
- Sempou, E.; Biasini, E.; Pinzon-Olejua, A.; Harris, D.A.; Malaga-Trillo, E. Activation of zebrafish src family kinases by the prion protein is an amyloid-beta-sensitive signal that prevents the endocytosis and degradation of e-cadherin/beta-catenin complexes in vivo. Mol. Neurodegener. 2016, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Huc-Brandt, S.; Hieu, N.; Imberdis, T.; Cubedo, N.; Silhol, M.; Leighton, P.L.; Domaschke, T.; Allison, W.T.; Perrier, V.; Rossel, M. Zebrafish prion protein prp2 controls collective migration process during lateral line sensory system development. PLoS ONE 2014, 9, e113331. [Google Scholar] [CrossRef] [PubMed]
- Pantera, B.; Bini, C.; Cirri, P.; Paoli, P.; Camici, G.; Manao, G.; Caselli, A. Prpc activation induces neurite outgrowth and differentiation in pc12 cells: Role for caveolin-1 in the signal transduction pathway. J. Neurochem. 2009, 110, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor ncam to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beraldo, F.H.; Arantes, C.P.; Santos, T.G.; Machado, C.F.; Roffe, M.; Hajj, G.N.; Lee, K.S.; Magalhaes, A.C.; Caetano, F.A.; Mancini, G.L.; et al. Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin gamma1 chain. FASEB J. 2011, 25, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Emsley, J.G.; Ozdinler, P.H.; Lindquist, S.; Macklis, J.D. Prion protein (prpc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 3416–3421. [Google Scholar] [CrossRef] [PubMed]
- Watt, N.T.; Taylor, D.R.; Kerrigan, T.L.; Griffiths, H.H.; Rushworth, J.V.; Whitehouse, I.J.; Hooper, N.M. Prion protein facilitates uptake of zinc into neuronal cells. Nat. Commun. 2012, 3, 1134. [Google Scholar] [CrossRef] [PubMed]
- Pushie, M.J.; Pickering, I.J.; Martin, G.R.; Tsutsui, S.; Jirik, F.R.; George, G.N. Prion protein expression level alters regional copper, iron and zinc content in the mouse brain. Metallomics 2011, 3, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Zanata, S.M.; Lopes, M.H.; Mercadante, A.F.; Hajj, G.N.; Chiarini, L.B.; Nomizo, R.; Freitas, A.R.; Cabral, A.L.; Lee, K.S.; Juliano, M.A.; et al. Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. EMBO J. 2002, 21, 3307–3316. [Google Scholar] [CrossRef] [PubMed]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Doeppner, T.R.; Kaltwasser, B.; Schlechter, J.; Jaschke, J.; Kilic, E.; Bahr, M.; Hermann, D.M.; Weise, J. Cellular prion protein promotes post-ischemic neuronal survival, angioneurogenesis and enhances neural progenitor cell homing via proteasome inhibition. Cell Death Dis. 2015, 6, e2024. [Google Scholar] [CrossRef] [PubMed]
- Sakurai-Yamashita, Y.; Sakaguchi, S.; Yoshikawa, D.; Okimura, N.; Masuda, Y.; Katamine, S.; Niwa, M. Female-specific neuroprotection against transient brain ischemia observed in mice devoid of prion protein is abolished by ectopic expression of prion protein-like protein. Neuroscience 2005, 136, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Khosravani, H.; Zhang, Y.F.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.N.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting nmda receptors. J. Cell Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Mercer, R.C.; Ma, L.; Watts, J.C.; Strome, R.; Wohlgemuth, S.; Yang, J.; Cashman, N.R.; Coulthart, M.B.; Schmitt-Ulms, G.; Jhamandas, J.H.; et al. The prion protein modulates a-type K+ currents mediated by kv4.2 complexes through dipeptidyl aminopeptidase-like protein 6. J. Biol. Chem. 2013, 288, 37241–37255. [Google Scholar] [CrossRef] [PubMed]
- Criado, J.R.; Sanchez-Alavez, M.; Conti, B.; Giacchino, J.L.; Wills, D.N.; Henriksen, S.J.; Race, R.; Manson, J.C.; Chesebro, B.; Oldstone, M.B. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of prp in neurons. Neurobiol. Dis. 2005, 19, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Coitinho, A.S.; Roesler, R.; Martins, V.R.; Brentani, R.R.; Izquierdo, I. Cellular prion protein ablation impairs behavior as a function of age. Neuroreport 2003, 14, 1375–1379. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Greis, C.; Ottis, P.; Silva, C.J.; Schulz-Schaeffer, W.J.; Wrede, A.; Koppe, K.; Onisko, B.; Requena, J.R.; Govindarajan, N.; et al. Loss of prion protein leads to age-dependent behavioral abnormalities and changes in cytoskeletal protein expression. Mol. Neurobiol. 2014, 50, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.L.A.; Nadolski, N.; Morill, A.; Hamilton, T.J.; Allison, W.T. Prion protein contributes to learning and memory in zebrafish. Biol. Open 2017. in revision. [Google Scholar]
- Lauren, J. Cellular prion protein as a therapeutic target in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 227–244. [Google Scholar] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Salazar, S.V.; Strittmatter, S.M. Cellular prion protein as a receptor for amyloid-beta oligomers in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Um, J.W.; Kaufman, A.C.; Kostylev, M.; Heiss, J.K.; Stagi, M.; Takahashi, H.; Kerrisk, M.E.; Vortmeyer, A.; Wisniewski, T.; Koleske, A.J.; et al. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 2013, 79, 887–902. [Google Scholar] [CrossRef] [PubMed]
- Yehiely, F.; Bamborough, P.; Da Costa, M.; Perry, B.J.; Thinakaran, G.; Cohen, F.E.; Carlson, G.A.; Prusiner, S.B. Identification of candidate proteins binding to prion protein. Neurobiol. Dis. 1997, 3, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Ulms, G.; Hansen, K.; Liu, J.; Cowdrey, C.; Yang, J.; DeArmond, S.J.; Cohen, F.E.; Prusiner, S.B.; Baldwin, M.A. Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat. Biotechnol. 2004, 22, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Markham, K.; Chen, F.; Weerasekera, R.; Watts, J.; Horne, P.; Wakutani, Y.; Bagshaw, R.; Mathews, P.M.; Fraser, P.E.; et al. The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteom. 2008, 7, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, I.J.; Miners, J.S.; Glennon, E.B.; Kehoe, P.G.; Love, S.; Kellett, K.A.; Hooper, N.M. Prion protein is decreased in Alzheimer’s brain and inversely correlates with bace1 activity, amyloid-beta levels and braak stage. PLoS ONE 2013, 8, e59554. [Google Scholar] [CrossRef] [PubMed]
- Whitehouse, I.J.; Brown, D.; Baybutt, H.; Diack, A.B.; Kellett, K.A.; Piccardo, P.; Manson, J.C.; Hooper, N.M. Ablation of prion protein in wild type human amyloid precursor protein (app) transgenic mice does not alter the proteolysis of app, levels of amyloid-beta or pathologic phenotype. PLoS ONE 2016, 11, e0159119. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free. Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
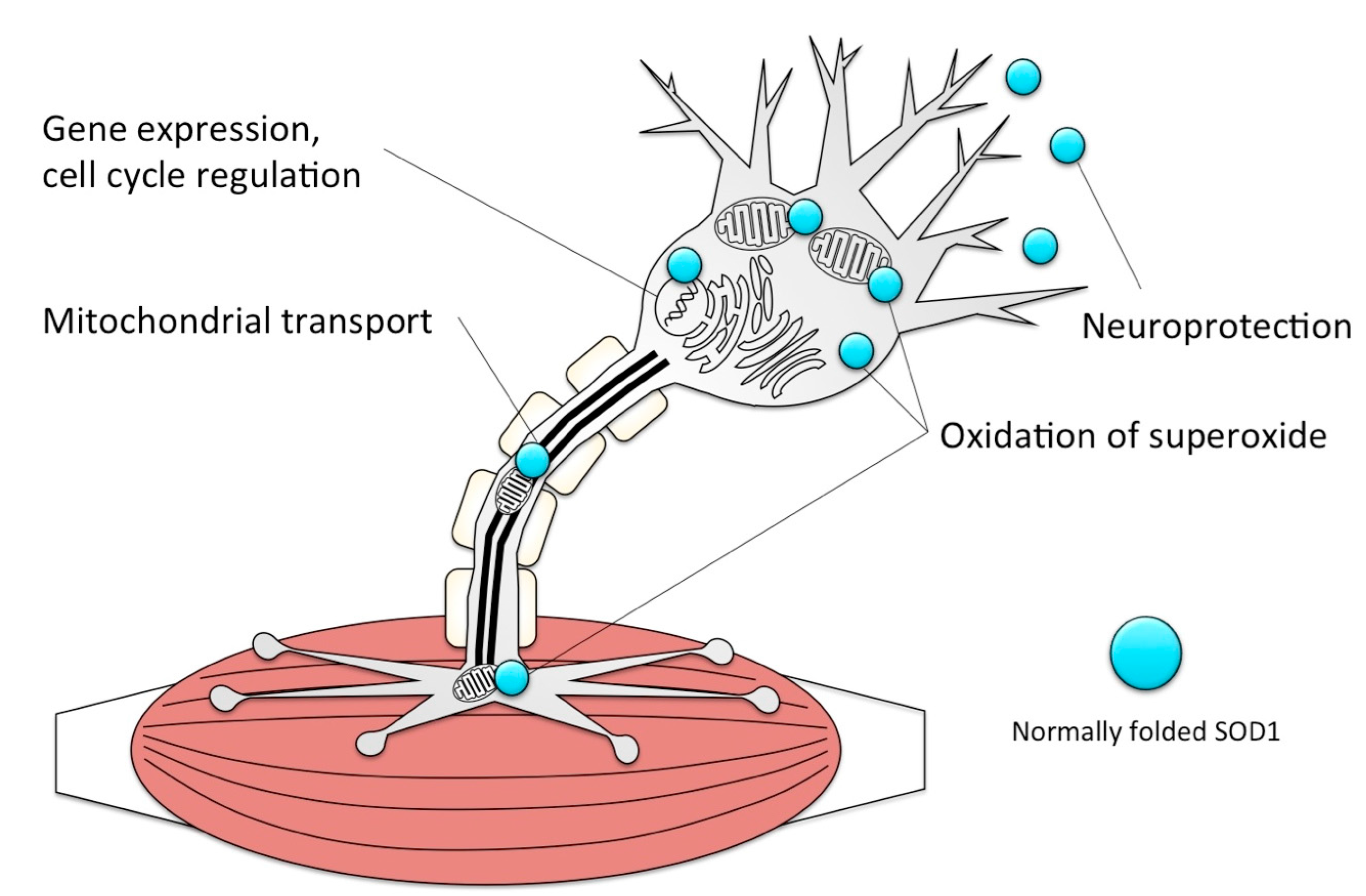
- Bendotti, C.; Carri, M.T. Lessons from models of sod1-linked familial als. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Harraz, M.M.; Marden, J.J.; Zhou, W.; Zhang, Y.; Williams, A.; Sharov, V.S.; Nelson, K.; Luo, M.; Paulson, H.; Schoneich, C.; et al. Sod1 mutations disrupt redox-sensitive rac regulation of nadph oxidase in a familial als model. J. Clin. Investig. 2008, 118, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Amyotrophic Lateral Sclerosis Online Genetics Database. Available online: http://alsod.iop.kcl.ac.uk/home.aspx (accessed on 1 July 2017).
- Grad, L.I.; Fernando, S.M.; Cashman, N.R. From molecule to molecule and cell to cell: Prion-like mechanisms in amyotrophic lateral sclerosis. Neurobiol. Dis. 2015, 77, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Bidhendi, E.E.; Bergh, J.; Zetterstrom, P.; Andersen, P.M.; Marklund, S.L.; Brannstrom, T. Two superoxide dismutase prion strains transmit amyotrophic lateral sclerosis-like disease. J. Clin. Investig. 2016, 126, 2249–2253. [Google Scholar] [CrossRef] [PubMed]
- Munch, C.; O’Brien, J.; Bertolotti, A. Prion-like propagation of mutant superoxide dismutase-1 misfolding in neuronal cells. Proc. Natl. Acad. Sci. USA. 2011, 108, 3548–3553. [Google Scholar] [CrossRef] [PubMed]
- Prudencio, M.; Durazo, A.; Whitelegge, J.P.; Borchelt, D.R. An examination of wild-type SOD1 in modulating the toxicity and aggregation of als-associated mutant sod1. Hum. Mol. Genet. 2010, 19, 4774–4789. [Google Scholar] [CrossRef] [PubMed]
- Witan, H.; Gorlovoy, P.; Kaya, A.M.; Koziollek-Drechsler, I.; Neumann, H.; Behl, C.; Clement, A.M. Wild-type cu/zn superoxide dismutase (SOD1) does not facilitate, but impedes the formation of protein aggregates of amyotrophic lateral sclerosis causing mutant sod1. Neurobiol. Dis. 2009, 36, 331–342. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, V.; Walker, A.K.; Yerbury, J.; Soo, K.Y.; Farg, M.A.; Hoang, V.; Zeineddine, R.; Spencer, D.; Atkin, J.D. Extracellular wildtype and mutant SOD1 induces ER-GOLGI pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell. Mol. Life Sci. 2013, 70, 4181–4195. [Google Scholar] [CrossRef] [PubMed]
- Guest, W.C.; Silverman, J.M.; Pokrishevsky, E.; O’Neill, M.A.; Grad, L.I.; Cashman, N.R. Generalization of the prion hypothesis to other neurodegenerative diseases: An imperfect fit. J. Toxicol. Environ. Health. A 2011, 74, 1433–1459. [Google Scholar] [CrossRef] [PubMed]
- Bosco, D.A.; LaVoie, M.J.; Petsko, G.A.; Ringe, D. Proteostasis and movement disorders: Parkinson’s disease and amyotrophic lateral sclerosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a007500. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, T.E.; Li, Y.; Cooper, D.; Gearing, M.; Julien, J.P.; Rothstein, J.D.; Boylan, K.; Glass, J.D. Localization of a toxic form of superoxide dismutase 1 protein to pathologically affected tissues in familial als. Proc. Natl. Acad. Sci. USA 2012, 109, 5505–5510. [Google Scholar] [CrossRef] [PubMed]
- Weser, U.; Miesel, R.; Hartmann, H.J. Mummified enzymes. Nature 1989, 341, 696. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, A. Propagation and replication of misfolded sod1: Implications for amyotrophic lateral sclerosis. In Proteopathic Seeds and Neurodegenerative Diseases; Jucker, M., Christen, Y., Eds.; Springer Science: Berlin, Germany, 2013; pp. 115–122. [Google Scholar]
- Malinowski, D.P.; Fridovich, I. Subunit association and side-chain reactivities of bovine erythrocyte superoxide dismutase in denaturing solvents. Biochemistry 1979, 18, 5055–5060. [Google Scholar] [CrossRef] [PubMed]
- Saccon, R.A.; Bunton-Stasyshyn, R.K.; Fisher, E.M.; Fratta, P. Is SOD1 loss of function involved in amyotrophic lateral sclerosis? Brain 2013, 136, 2342–2358. [Google Scholar] [CrossRef] [PubMed]
- Reaume, A.G.; Elliott, J.L.; Hoffman, E.K.; Kowall, N.W.; Ferrante, R.J.; Siwek, D.F.; Wilcox, H.M.; Flood, D.G.; Beal, M.F.; Brown, R.H., Jr.; et al. Motor neurons in cu/zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat. Genet. 1996, 13, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Reaume, A.G.; Huang, T.T.; Carlson, E.; Murakami, K.; Chen, S.F.; Hoffman, E.K.; Scott, R.W.; Epstein, C.J.; Chan, P.H. Reduction of cuzn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J. Neurosci. 1997, 17, 4180–4189. [Google Scholar] [PubMed]
- Ivannikov, M.V.; Van Remmen, H. Sod1 gene ablation in adult mice leads to physiological changes at the neuromuscular junction similar to changes that occur in old wild-type mice. Free. Radic. Biol. Med. 2015, 84, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ivannikov, M.V.; Walsh, M.E.; Liu, Y.; Zhang, Y.; Jaramillo, C.A.; Macleod, G.T.; Van Remmen, H. The lack of cuznsod leads to impaired neurotransmitter release, neuromuscular junction destabilization and reduced muscle strength in mice. PLoS ONE 2014, 9, e100834. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Li, Y.; Asress, S.A.; Jones, D.P.; Glass, J.D. Absence of sod1 leads to oxidative stress in peripheral nerve and causes a progressive distal motor axonopathy. Exp. Neurol. 2012, 233, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Song, W.; Liu, Y.; Chaudhuri, A.; Pieke-Dahl, S.; Strong, R.; Huang, T.T.; Epstein, C.J.; Roberts, L.J., 2nd; Csete, M.; et al. Absence of cuzn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free. Radic. Biol. Med. 2006, 40, 1993–2004. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Van Blitterswijk, M.; Gulati, S.; Smoot, E.; Jaffa, M.; Maher, N.; Hyman, B.T.; Ivinson, A.J.; Scherzer, C.R.; Schoenfeld, D.A.; Cudkowicz, M.E.; et al. Anti-superoxide dismutase antibodies are associated with survival in patients with sporadic amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of oxidative damage on the stability and dimerization of superoxide dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- White, M.A.; Sreedharan, J. Amyotrophic lateral sclerosis: Recent genetic highlights. Curr. Opin. Neurol. 2016, 29, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Hashizume, K.; Hirasawa, M.; Imamura, Y.; Noda, S.; Shimizu, T.; Shinoda, K.; Kurihara, T.; Noda, K.; Ozawa, Y.; Ishida, S.; et al. Retinal dysfunction and progressive retinal cell death in sod1-deficient mice. Am. J. Pathol. 2008, 172, 1325–1331. [Google Scholar] [CrossRef] [PubMed]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in sod1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Ozawa, Y.; Yoshida, T.; Kurihara, T.; Hirasawa, M.; Ozeki, N.; Shiba, D.; Noda, K.; Ishida, S.; Tsubota, K. Retinal ganglion cell loss in superoxide dismutase 1 deficiency. Investig. Ophthalmol. Vis. Sci. 2011, 52, 4143–4150. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; van der Merwe, J.; Kim, C.; Haldiman, T.; McKenzie, D.; Safar, J.G.; Westaway, D. Prion infectivity plateaus and conversion to symptomatic disease originate from falling precursor levels and increased oligomeric prpsc species. J. Virol. 2015, 89, 12418–12426. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Thune, K.; Schmitz, M.; Ansoleaga, B.; Frau-Mendez, M.A.; Cramm, M.; Tahir, W.; Gotzmann, N.; Berjaoui, S.; Carmona, M.; et al. Identification of new molecular alterations in fatal familial insomnia. Hum. Mol. Genet. 2016, 25, 2417–2436. [Google Scholar] [CrossRef] [PubMed]
- Jan, J.E.; Reiter, R.J.; Wasdell, M.B.; Bax, M. The role of the thalamus in sleep, pineal melatonin production, and circadian rhythm sleep disorders. J. Pineal Res. 2009, 46, 1–7. [Google Scholar] [CrossRef] [PubMed]
- DelRosso, L.M.; Hoque, R. The cerebellum and sleep. Neurol. Clin. 2014, 32, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hammer, N.D.; Wang, X.; McGuffie, B.A.; Chapman, M.R. Amyloids: Friend or foe? J. Alzheimers Dis. 2008, 13, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Badtke, M.P.; Hammer, N.D.; Chapman, M.R. Functional amyloids signal their arrival. Sci. Signal. 2009, 2, pe43. [Google Scholar] [CrossRef] [PubMed]
- Fowler, D.M.; Koulov, A.V.; Alory-Jost, C.; Marks, M.S.; Balch, W.E.; Kelly, J.W. Functional amyloid formation within mammalian tissue. PLoS Biol. 2006, 4, e6. [Google Scholar] [CrossRef] [PubMed]
- Maji, S.K.; Perrin, M.H.; Sawaya, M.R.; Jessberger, S.; Vadodaria, K.; Rissman, R.A.; Singru, P.S.; Nilsson, K.P.; Simon, R.; Schubert, D.; et al. Functional amyloids as natural storage of peptide hormones in pituitary secretory granules. Science 2009, 325, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The rip1/rip3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Nukina, N. Functional diversity of protein fibrillar aggregates from physiology to rna granules to neurodegenerative diseases. Biochim. Biophys. Acta 2013, 1832, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Wasmeier, C.; Hume, A.N.; Bolasco, G.; Seabra, M.C. Melanosomes at a glance. J. Cell. Sci. 2008, 121, 3995–3999. [Google Scholar] [CrossRef] [PubMed]
- Rochin, L.; Hurbain, I.; Serneels, L.; Fort, C.; Watt, B.; Leblanc, P.; Marks, M.S.; De Strooper, B.; Raposo, G.; van Niel, G. Bace2 processes pmel to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10658–10663. [Google Scholar] [CrossRef] [PubMed]
- Brunberg, E.; Andersson, L.; Cothran, G.; Sandberg, K.; Mikko, S.; Lindgren, G. A missense mutation in pmel17 is associated with the silver coat color in the horse. BMC Genet. 2006, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kerje, S.; Sharma, P.; Gunnarsson, U.; Kim, H.; Bagchi, S.; Fredriksson, R.; Schutz, K.; Jensen, P.; von Heijne, G.; Okimoto, R.; et al. The dominant white, dun and smoky color variants in chicken are associated with insertion/deletion polymorphisms in the pmel17 gene. Genetics 2004, 168, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.S.; Wilbe, M.; Viluma, A.; Cothran, G.; Ekesten, B.; Ewart, S.; Lindgren, G. Equine multiple congenital ocular anomalies and silver coat colour result from the pleiotropic effects of mutant pmel. PLoS ONE 2013, 8, e75639. [Google Scholar] [CrossRef] [PubMed]
- Schmutz, S.M.; Dreger, D.L. Interaction of mc1r and pmel alleles on solid coat colors in highland cattle. Anim. Genet. 2013, 44, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, C.; Weikard, R. An investigation into the genetic background of coat colour dilution in a charolais x german holstein F2 resource population. Anim. Genet. 2007, 38, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Kwon, B.S.; Halaban, R.; Ponnazhagan, S.; Kim, K.; Chintamaneni, C.; Bennett, D.; Pickard, R.T. Mouse silver mutation is caused by a single base insertion in the putative cytoplasmic domain of pmel 17. Nucleic Acids Res. 1995, 23, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.A.; Wahl, J.M.; Rees, C.A.; Murphy, K.E. Retrotransposon insertion in silv is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. USA 2006, 103, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Schonthaler, H.B.; Lampert, J.M.; von Lintig, J.; Schwarz, H.; Geisler, R.; Neuhauss, S.C. A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev. Biol. 2005, 284, 421–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watt, B.; Tenza, D.; Lemmon, M.A.; Kerje, S.; Raposo, G.; Andersson, L.; Marks, M.S. Mutations in or near the transmembrane domain alter pmel amyloid formation from functional to pathogenic. PLoS Genet. 2011, 7, e1002286. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.R.; Watt, B.; Fard, S.S.; Tenza, D.; Mannstrom, P.; Narfstrom, K.; Ekesten, B.; Ito, S.; Wakamatsu, K.; Larsson, J.; et al. Inactivation of pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet. 2011, 7, e1002285. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.D.; Peles, D.; Wakamatsu, K.; Ito, S. Current challenges in understanding melanogenesis: Bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009, 22, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Watt, B.; van Niel, G.; Raposo, G.; Marks, M.S. Pmel: A pigment cell-specific model for functional amyloid formation. Pigment Cell Melanoma Res. 2013, 26, 300–315. [Google Scholar] [CrossRef] [PubMed]
- Seelig, D.M.; Goodman, P.A.; Skinner, P.J. Potential approaches for heterologous prion protein treatment of prion diseases. Prion 2016, 10, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.J., Jr.; Gajdusek, D.C. Experimental subacute spongiform virus encephalopathies in primates and other laboratory animals. Science 1973, 182, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Barlow, R.M.; Rennie, J.C. The fate of ME7 scrapie infection in rats, guinea-pigs and rabbits. Res. Vet. Sci. 1976, 21, 110–111. [Google Scholar] [PubMed]
- Skinner, P.J.; Kim, H.O.; Bryant, D.; Kinzel, N.J.; Reilly, C.; Priola, S.A.; Ward, A.E.; Goodman, P.A.; Olson, K.; Seelig, D.M. Treatment of prion disease with heterologous prion proteins. PLoS ONE 2015, 10, e0131993. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; Zuccato, C.; Tartari, M. Normal huntingtin function: An alternative approach to huntington’s disease. Nat. Rev. Neurosci. 2005, 6, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Pimplikar, S.W. Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PLoS ONE 2012, 7, e34209. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.; Liang, J.O.; DiMonte, K.; Sullivan, J.; Pimplikar, S.W. Amyloid precursor protein is required for convergent-extension movements during zebrafish development. Dev. Biol. 2009, 335, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal prp in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871–874. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Espinoza, R.; Morales, R.; Concha-Marambio, L.; Moreno-Gonzalez, I.; Moda, F.; Soto, C. Treatment with a non-toxic, self-replicating anti-prion delays or prevents prion disease in vivo. Mol. Psychiatry 2017. [Google Scholar] [CrossRef] [PubMed]











© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allison, W.T.; DuVal, M.G.; Nguyen-Phuoc, K.; Leighton, P.L.A. Reduced Abundance and Subverted Functions of Proteins in Prion-Like Diseases: Gained Functions Fascinate but Lost Functions Affect Aetiology. Int. J. Mol. Sci. 2017, 18, 2223. https://doi.org/10.3390/ijms18102223
Allison WT, DuVal MG, Nguyen-Phuoc K, Leighton PLA. Reduced Abundance and Subverted Functions of Proteins in Prion-Like Diseases: Gained Functions Fascinate but Lost Functions Affect Aetiology. International Journal of Molecular Sciences. 2017; 18(10):2223. https://doi.org/10.3390/ijms18102223
Chicago/Turabian StyleAllison, W. Ted, Michèle G. DuVal, Kim Nguyen-Phuoc, and Patricia L. A. Leighton. 2017. "Reduced Abundance and Subverted Functions of Proteins in Prion-Like Diseases: Gained Functions Fascinate but Lost Functions Affect Aetiology" International Journal of Molecular Sciences 18, no. 10: 2223. https://doi.org/10.3390/ijms18102223
APA StyleAllison, W. T., DuVal, M. G., Nguyen-Phuoc, K., & Leighton, P. L. A. (2017). Reduced Abundance and Subverted Functions of Proteins in Prion-Like Diseases: Gained Functions Fascinate but Lost Functions Affect Aetiology. International Journal of Molecular Sciences, 18(10), 2223. https://doi.org/10.3390/ijms18102223