The Peptidylarginine Deiminase Inhibitor Cl-Amidine Suppresses Inducible Nitric Oxide Synthase Expression in Dendritic Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
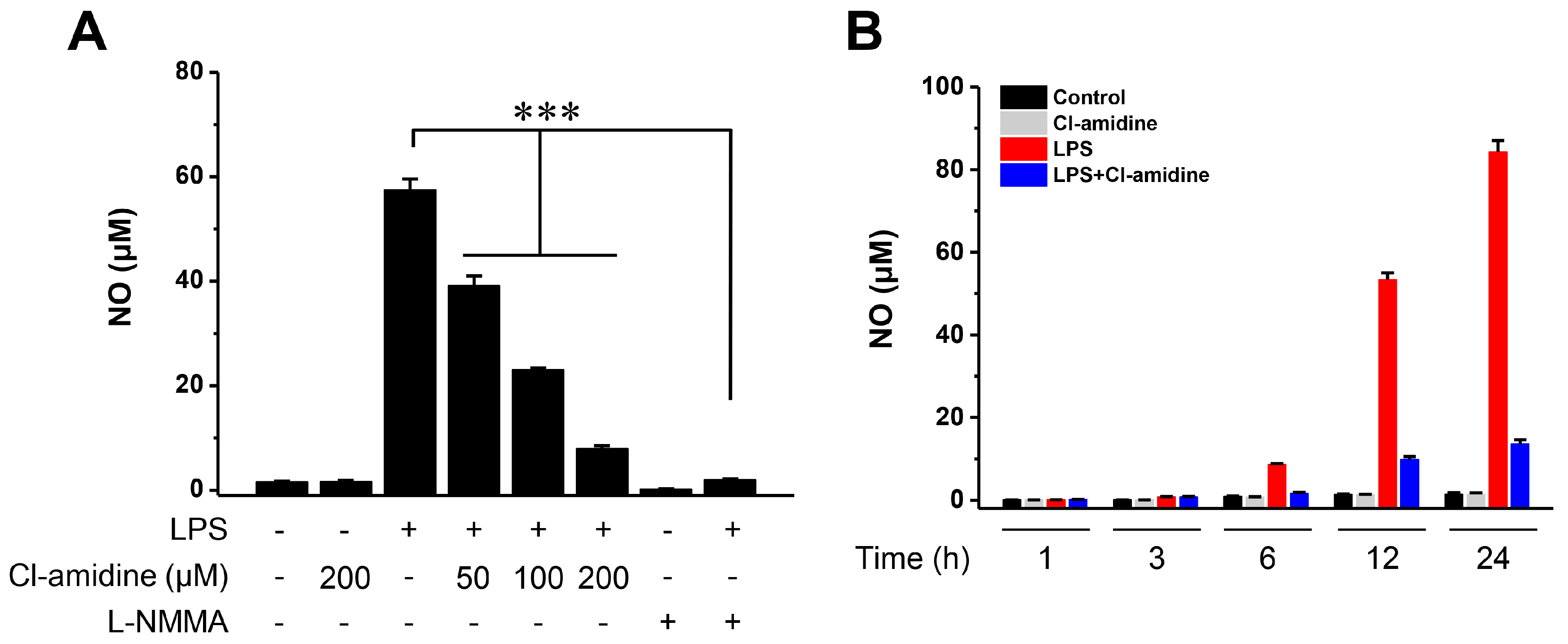
2.1. Cl-Amidine Suppresses NO Generation in Lipopolysaccharide-Stimulated Dendritic Cells
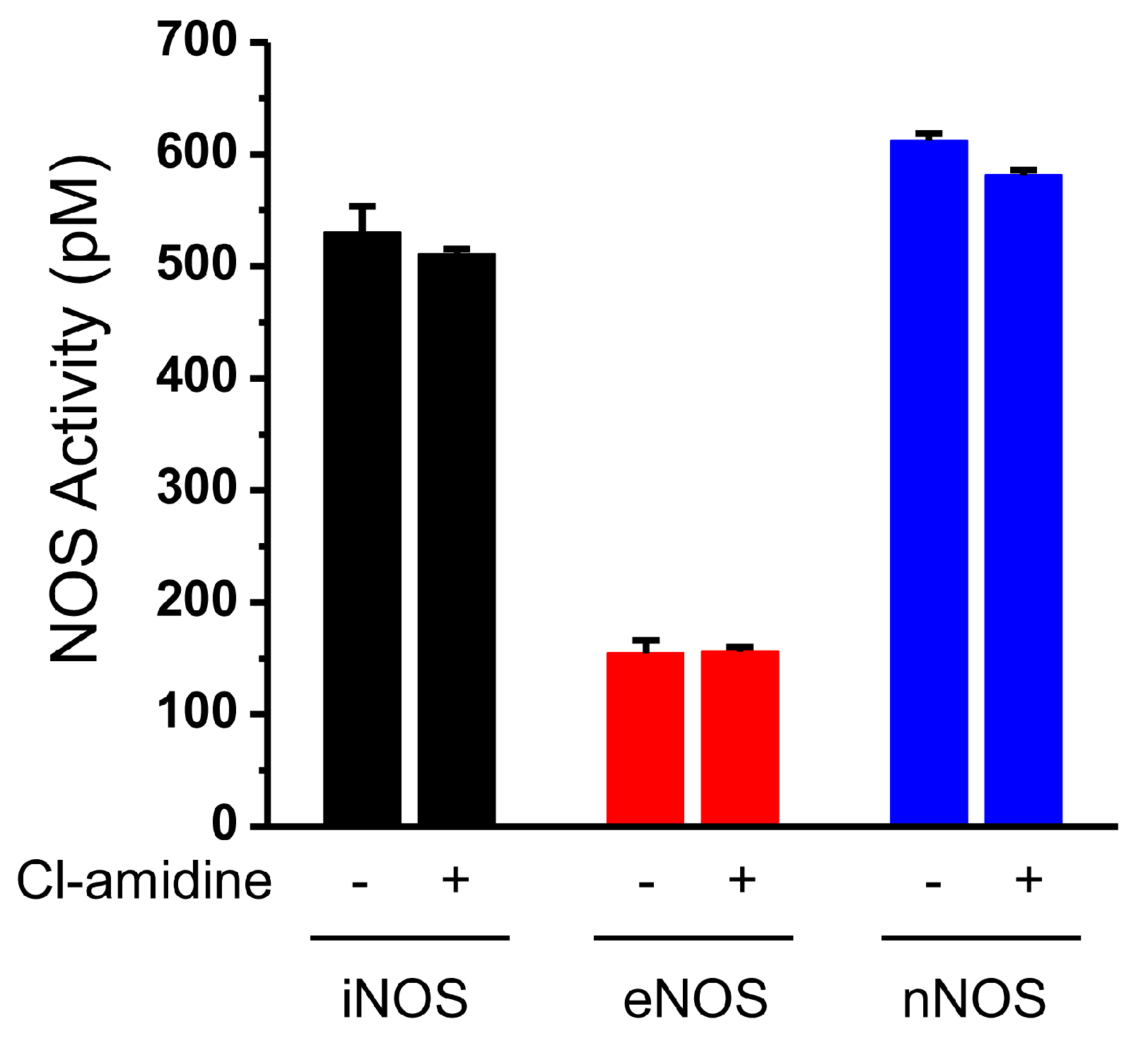
2.2. NOS Activity Is not Inhibited by Cl-Amidine
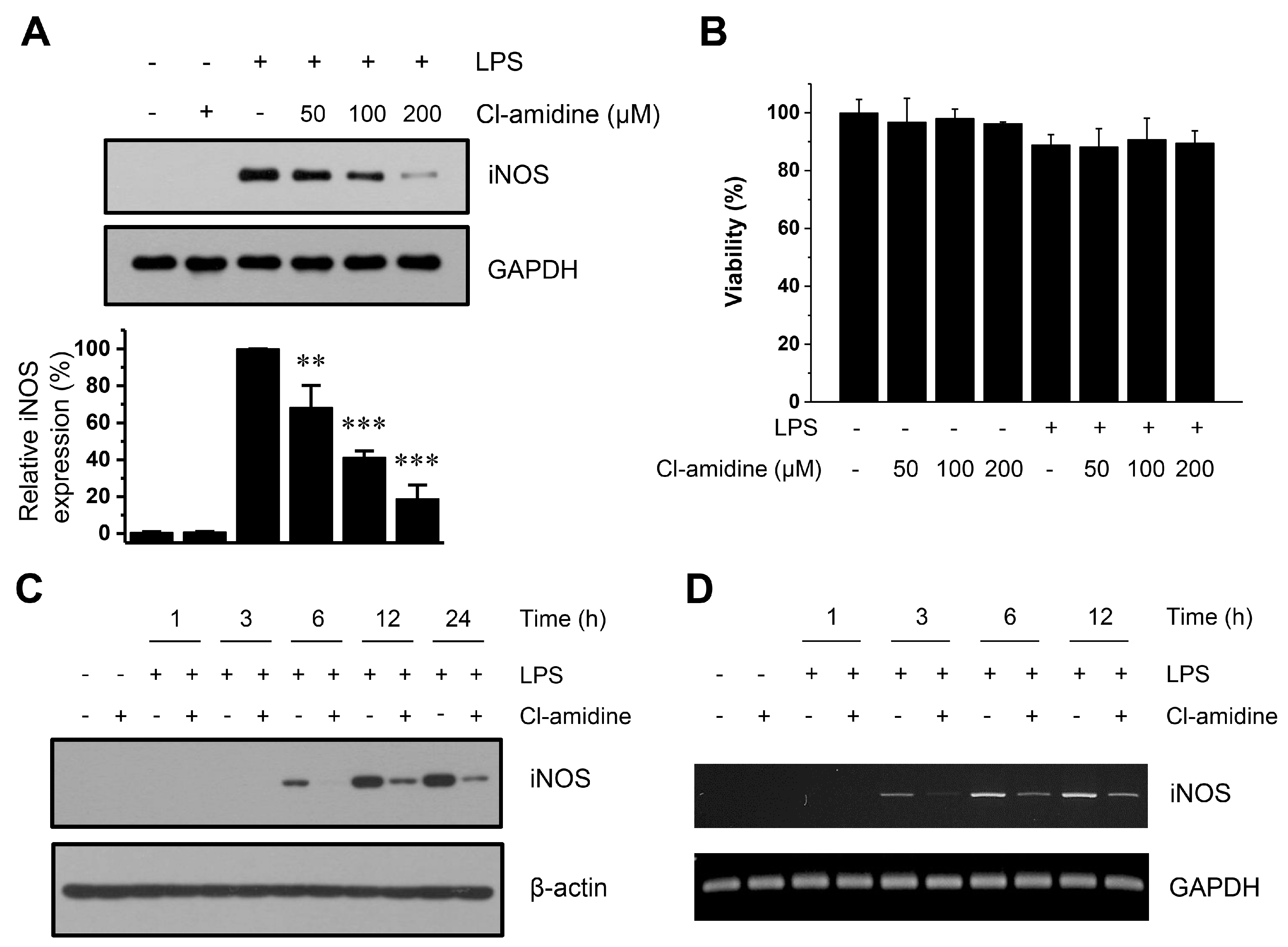
2.3. Cl-Amidine Suppresses iNOS Expression
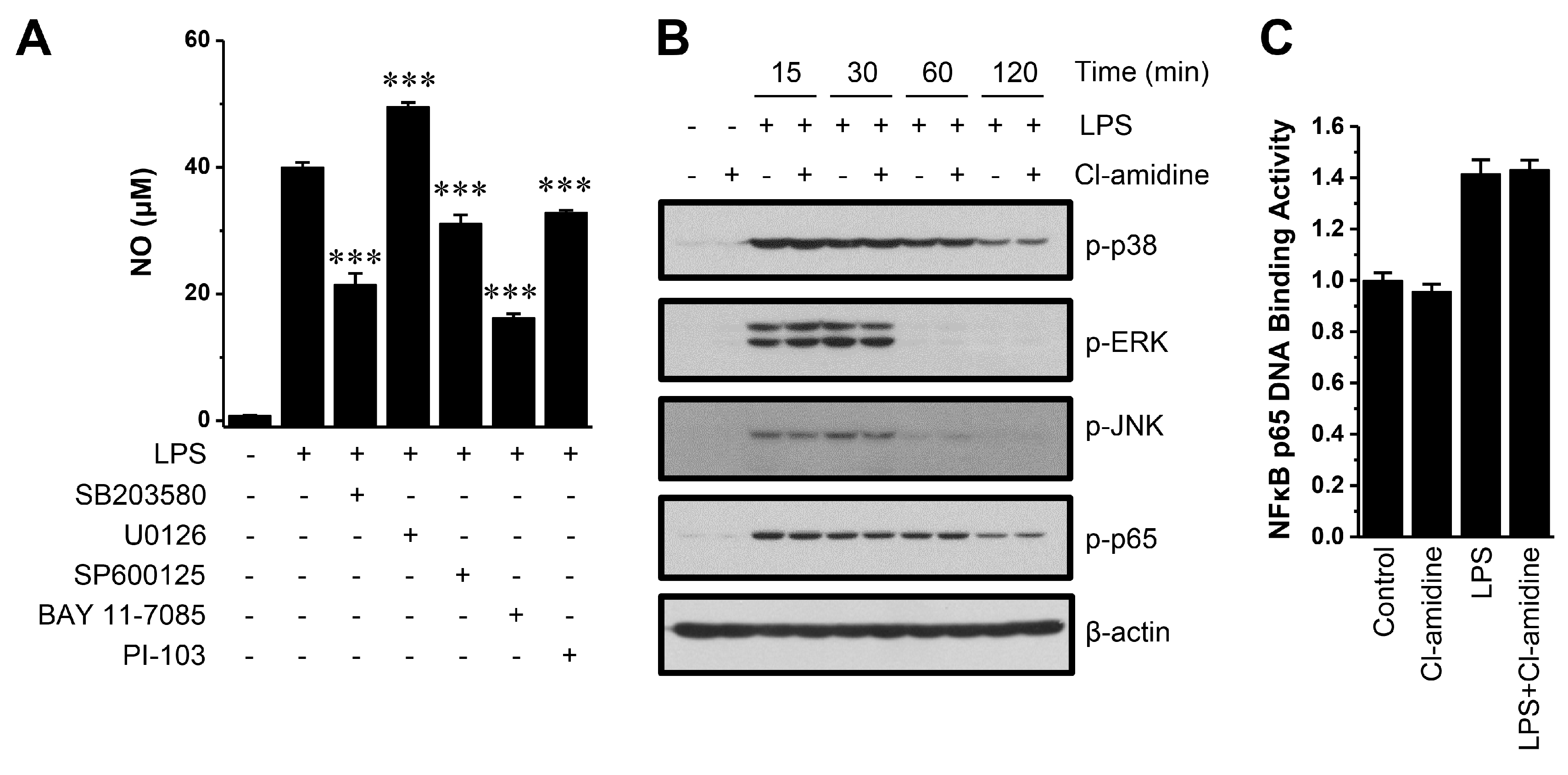
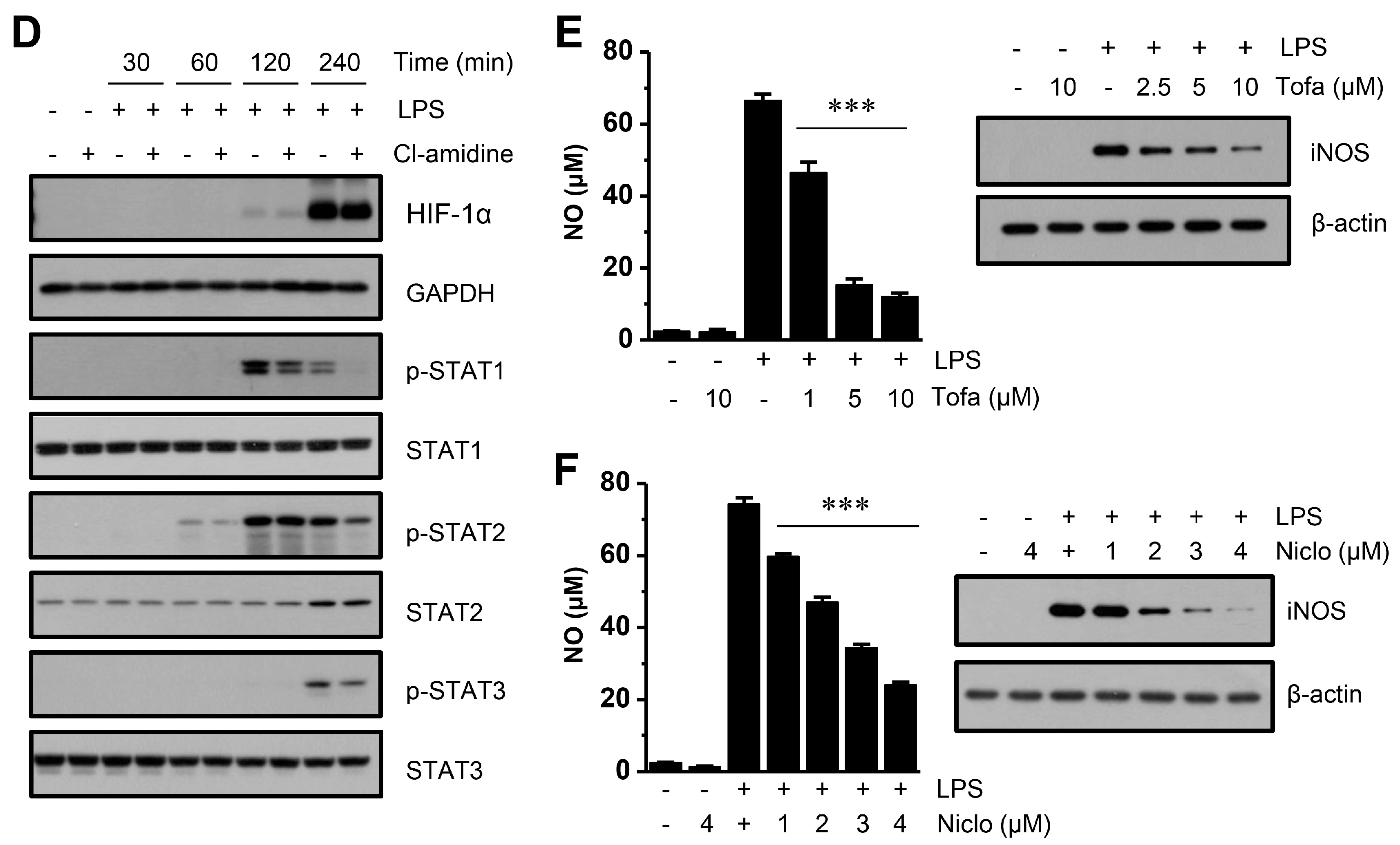
2.4. Cl-Amidine Does not Affect Mitogen-Activated Protein Kinases (MAPKs), Nuclear Factor-κB p65 (NF-κB p65) Signaling, or Hypoxia-Inducible Factor 1α (HIF-1α) Expression but Instead Attenuates STAT Activation
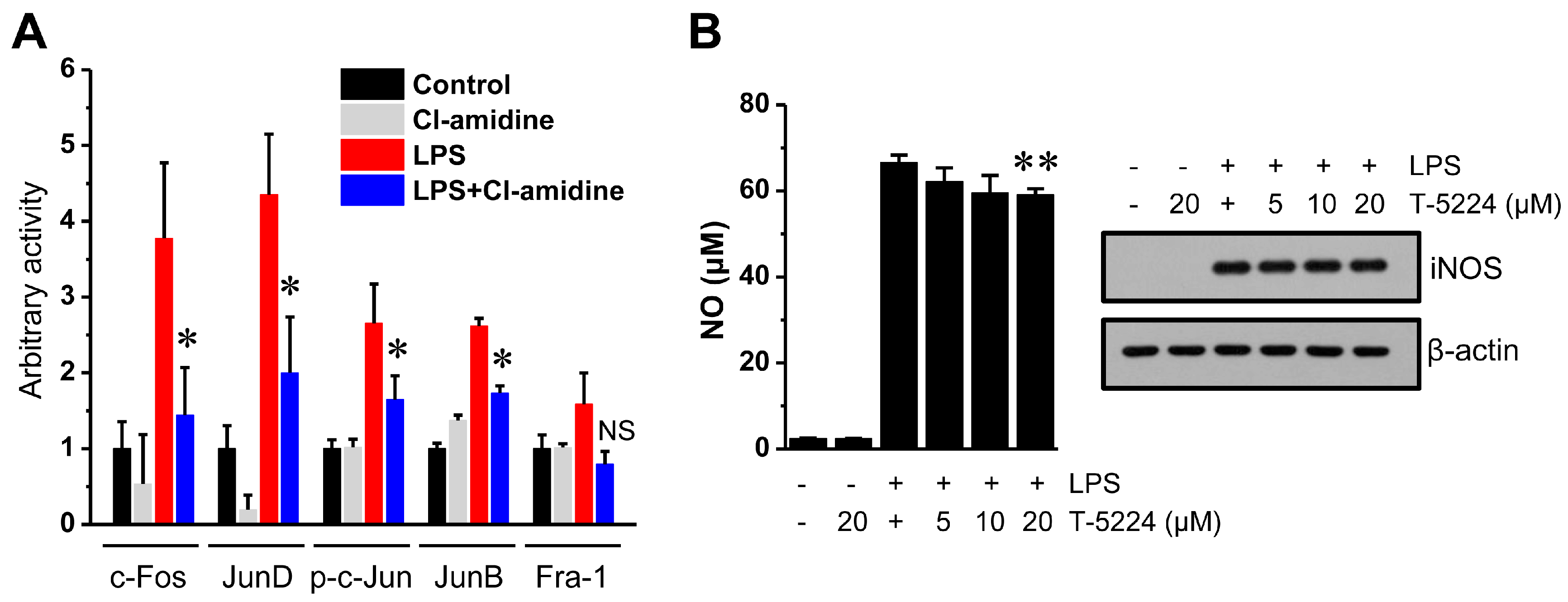
2.5. Cl-Amidine Impairs AP-1 Family Member Activity
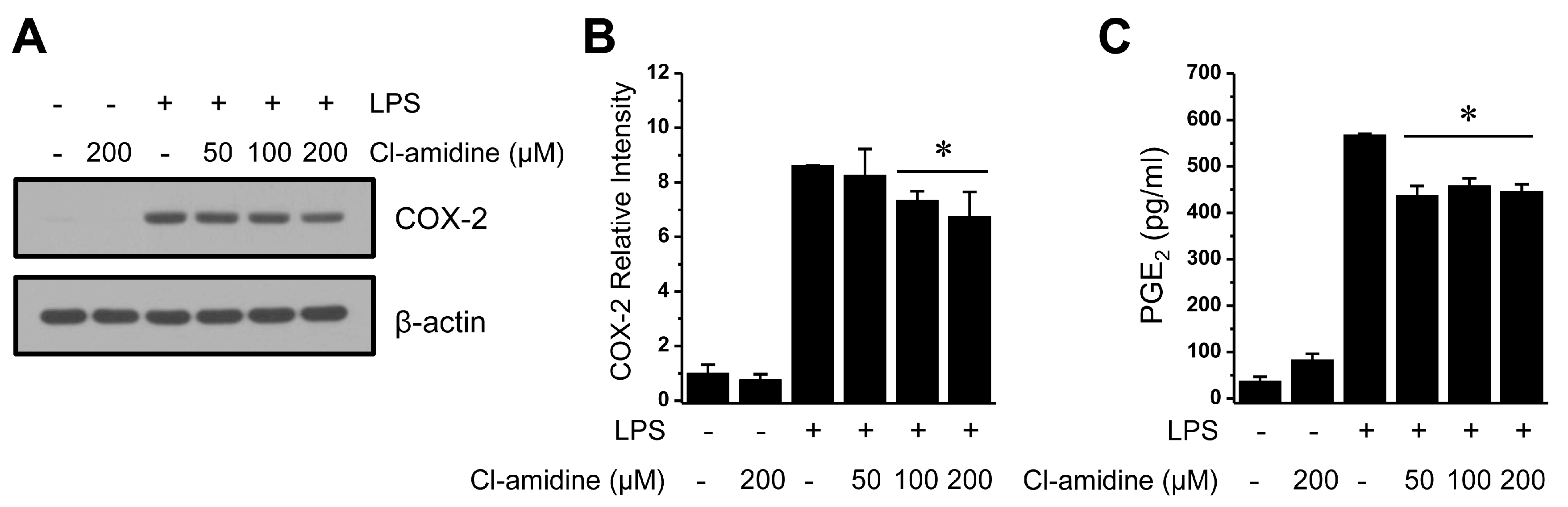
2.6. Ineffective Regulation of Cyclooxygenase 2 (COX-2) Expression and Prostaglandin E2 (PGE2) Production due to Cl-Amidine Treatment
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Culture
4.3. NO Detection and NOS Activity
4.4. Western Blotting Analysis
4.5. Semi-Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
4.6. NF-κB p65 Transcription Factor Activity Assay
4.7. Activator Protein-1 (AP-1) Transcription Activity Assay
4.8. Prostaglandin E2 (PGE2) Assay
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PAD | Pepitylarginine deiminase |
| iNOS | Inducible nitric oxide synthase |
| DC | Dendritic cell |
| LPS | Lipopolysaccharide |
| AP-1 | Activator protein 1 |
| MAPK | Mitogen-activated protein kinases |
| HIF-1 | Hypoxia-inducible factor 1 |
| NF-κB p65 | Nuclear factor-kappaB p65 |
| PI3K | Phosphatidylinositol 3-kinase |
| JNK | C-Jun N-terminal kinase |
| IL | Interleukin |
| COX-2 | Cyclooxygenase 2 |
| PGE2 | Prostaglandin E2 |
| JAK | Janus kinase |
| STAT | Signal transducer and activator of transcription |
References
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural basis for Ca2+-induced activation of human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. Bioessays 2003, 25, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.; Ishigami, A.; Maruyama, N.; Carp, R.I.; Kim, Y.S.; Choi, E.K. Peptidylarginine deiminase and protein citrullination in prion diseases: Strong evidence of neurodegeneration. Prion 2013, 7, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.E.; Thompson, P.R.; Hofseth, L.J. Protein Arginine Deiminases and Associated Citrullination: Physiological Functions and Diseases Associated with Dysregulation. Curr. Drug Targets 2015, 16, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Conigliaro, P.; Chimenti, M.S.; Triggianese, P.; Sunzini, F.; Novelli, L.; Perricone, C.; Perricone, R. Autoantibodies in inflammatory arthritis. Autoimmun. Rev. 2016, 15, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derksen, V.F.A.M.; Huizinga, T.W.J.; van der Woude, D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin. Immunopathol. 2017, 39, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Willis, V.C.; Gizinski, A.M.; Banda, N.K.; Causey, C.P.; Knuckley, B.; Cordova, K.N.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-l-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Moscarello, M.A.; Lei, H.; Mastronardi, F.G.; Winer, S.; Tsui, H.; Li, Z.; Ackerley, C.; Zhang, L.; Raijmakers, R.; Wood, D.D. Inhibition of peptidyl-arginine deiminases reverses protein-hypercitrullination and disease in mouse models of multiple sclerosis. Dis. Model. Mech. 2013, 6, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Chumanevich, A.A.; Causey, C.P.; Knuckley, B.A.; Jones, J.E.; Poudyal, D.; Chumanevich, A.P.; Davis, T.; Matesic, L.E.; Thompson, P.R.; Hofseth, L.J. Suppression of colitis in mice by Cl-amidine: A novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G929–G938. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.E.; Cui, X.; Causey, C.P.; Thompson, P.R.; Hofseth, L.J. Molecular targeting of protein arginine deiminases to suppress colitis and prevent colon cancer. Oncotarget 2015, 6, 36053–36062. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef] [PubMed]
- Ham, A.; Rabadi, M.; Kim, M.; Brown, K.M.; Ma, Z.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2014, 307, F1052–F1062. [Google Scholar] [CrossRef] [PubMed]
- Lange, S.; Rocha-Ferreira, E.; Thei, L.; Mawjee, P.; Bennett, K.; Thompson, P.R.; Subramanian, V.; Nicholas, A.P.; Peebles, D.; Hristova, M.; et al. Peptidylarginine deiminases: Novel drug targets for prevention of neuronal damage following hypoxic ischemic insult (HI) in neonates. J. Neurochem. 2014, 130, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, P.; Wang, S.; Hu, J.; Chen, X.A.; Wu, J.; Fisher, M.; Oshaben, K.; Zhao, N.; Gu, Y.; et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J. Biol. Chem. 2012, 287, 25941–25953. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef] [PubMed]
- Jang, B.; Kim, H.W.; Kim, J.S.; Kim, W.S.; Lee, B.R.; Kim, S.; Kim, H.; Han, S.J.; Ha, S.J.; Shin, S.J. Peptidylarginine deiminase inhibition impairs Toll-like receptor agonist-induced functional maturation of dendritic cells, resulting in the loss of T cell-proliferative capacity: A partial mechanism with therapeutic potential in inflammatory settings. J. Leukoc. Biol. 2015, 97, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Corinti, S.; Pastore, S.; Mascia, F.; Girolomoni, G. Regulatory role of nitric oxide on monocyte-derived dendritic cell functions. J. Interferon Cytokine Res. 2003, 23, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Clark, J.M.; Buzás, E.I.; Gorman, C.L.; Cope, A.P. Nitric oxide, chronic inflammation and autoimmunity. Immunol. Lett. 2007, 111, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric oxide synthase inhibition and oxidative stress in cardiovascular diseases: Possible therapeutic targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, C. Nitric oxide synthase in innate and adaptive immunity: An update. Trends Immunol. 2015, 36, 161–178. [Google Scholar] [CrossRef] [PubMed]
- Kleinert, H.; Pautz, A.; Linker, K.; Schwarz, P.M. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 2004, 500, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Suzuki, H.; Oda, H.; Akaike, T.; Azuma, Y.; Murakami, T.; Sugi, K.; Ito, T.; Ichinose, H.; Koyasu, S.; et al. Phosphoinositide 3-kinase in nitric oxide synthesis in macrophage: Critical dimerization of inducible nitric-oxide synthase. J. Biol. Chem. 2006, 281, 17736–17742. [Google Scholar] [CrossRef] [PubMed]
- Misko, T.P.; Trotter, J.L.; Cross, A.H. Mediation of inflammation by encephalitogenic cells: Interferon gamma induction of nitric oxide synthase and cyclooxygenase 2. J. Neuroimmunol. 1995, 61, 195–204. [Google Scholar] [CrossRef]
- Appleton, I.; Tomlinson, A.; Willoughby, D.A. Induction of cyclo-oxygenase and nitric oxide synthase in inflammation. Adv. Pharmacol. 1996, 35, 27–78. [Google Scholar] [PubMed]
- Kim, S.F.; Huri, D.A.; Snyder, S.H. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science 2005, 310, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.J.; Blake, D.R.; Palmer, R.M.; Moncada, S. Increased concentrations of nitrite in synovial fluid and serum samples suggest increased nitric oxide synthesis in rheumatic diseases. Ann. Rheum. Dis. 1992, 51, 1219–1222. [Google Scholar] [CrossRef] [PubMed]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytometry 2012, 81, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Benchabane, S.; Boudjelida, A.; Toumi, R.; Belguendouz, H.; Youinou, P.; Touil-Boukoffa, C. A case for IL-6, IL-17A, and nitric oxide in the pathophysiology of Sjögren’s syndrome. Int. J. Immunopathol. Pharmacol. 2016, 29, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Koncz, A.; Telarico, T.; Fernandez, D.; Ersek, B.; Buzás, E.; Perl, A. Central role of nitric oxide in the pathogenesis of rheumatoid arthritis and systemic lupus erythematosus. Arthritis Res. Ther. 2010, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.C.; Chuang, Y.H.; Tsai, Y.F.; Yu, H.P. Role of neutrophil extracellular traps following injury. Shock 2014, 41, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, Y.; Nakazawa, D.; Shida, H.; Hattanda, F.; Miyoshi, A.; Masuda, S.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production. Front. Immunol. 2016, 7, 227. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Kumar, S.; Jyoti, A.; Srinag, B.S.; Keshari, R.S.; Saluja, R.; Verma, A.; Mitra, K.; Barthwal, M.K.; Krishnamurthy, H.; et al. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide 2010, 22, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.B.; Kuiper, J.W.; Katchky, A.; Goldberg, H.; Glogauer, M. Rac2 is required for the formation of neutrophil extracellular traps. J. Leukoc. Biol. 2011, 90, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.L.; Chi, H.; Sun, L. Neutrophil Extracellular Traps of Cynoglossus semilaevis: Production Characteristics and Antibacterial Effect. Front. Immunol. 2017, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Andrade, F. A Critical Reappraisal of Neutrophil Extracellular Traps and NETosis Mimics Based on Differential Requirements for Protein Citrullination. Front. Immunol. 2016, 7, 461. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Hu, X.; Xu, H.; Wu, L.; Dai, Y.; Yang, L.; Xu, Z. Phosphatidylinositol 3-kinase inhibitor suppresses inducible nitric oxide synthase expression in bronchiole epithelial cells in asthmatic rats. Mol. Cell. Biochem. 2012, 359, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Guerra, M.J.; Castrillo, A.; Martín-Sanz, P.; Boscá, L. Negative regulation by phosphatidylinositol 3-kinase of inducible nitric oxide synthase expression in macrophages. J. Immunol. 1999, 162, 6184–6190. [Google Scholar] [PubMed]
- Koide, N.; Ito, H.; Mu, M.M.; Sugiyama, T.; Hassan, F.; Islam, S.; Mori, I.; Yoshida, T.; Yokochi, T. Inhibition of extracellular signal-regulated kinase 1/2 augments nitric oxide production in lipopolysaccharide-stimulated RAW264.7 macrophage cells. FEMS Immunol. Med. Microbiol. 2005, 45, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Dwivedi, N.; Bechtel, T.J.; Paulsen, J.L.; Muth, A.; Bawadekar, M.; Li, G.; Thompson, P.R.; Shelef, M.A.; Schiffer, C.A.; et al. Citrullination of NF-κB p65 promotes its nuclear localization and TLR-induced expression of IL-1β and TNFα. Sci. Immunol. 2017, 2, eaal3062. [Google Scholar] [CrossRef] [PubMed]
- Rabadi, M.; Kim, M.; D’Agati, V.; Lee, H.T. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am. J. Physiol. Ren. Physiol. 2016, 311, F437–F449. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.T.; Duarte, C.B.; Gonçalo, M.; Carvalho, A.P.; Lopes, M.C. Involvement of JAK2 and MAPK on type II nitric oxide synthase expression in skin-derived dendritic cells. Am. J. Physiol. 1999, 277, C1050–C1057. [Google Scholar] [PubMed]
- Stempelj, M.; Kedinger, M.; Augenlicht, L.; Klampfer, L. Essential role of the JAK/STAT1 signaling pathway in the expression of inducible nitric-oxide synthase in intestinal epithelial cells and its regulation by butyrate. J. Biol. Chem. 2007, 282, 9797–9804. [Google Scholar] [CrossRef] [PubMed]
- Dell’Albani, P.; Santangelo, R.; Torrisi, L.; Nicoletti, V.G.; de Vellis, J.; Giuffrida Stella, A.M. JAK/STAT signaling pathway mediates cytokine-induced iNOS expression in primary astroglial cell cultures. J. Neurosci. Res. 2001, 65, 417–424. [Google Scholar] [CrossRef] [PubMed]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer. 2010, 1, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H. Protein tyrosine nitration—An update. Arch. Biochem. Biophys. 2009, 484, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Benhar, M. Emerging Roles of Protein S-Nitrosylation in Macrophages and Cancer Cells. Curr. Med. Chem. 2016, 23, 2602–2617. [Google Scholar] [CrossRef] [PubMed]
- Fraszczak, J.; Trad, M.; Janikashvili, N.; Cathelin, D.; Lakomy, D.; Granci, V.; Morizot, A.; Audia, S.; Micheau, O.; Lagrost, L.; et al. Peroxynitrite-dependent killing of cancer cells and presentation of released tumor antigens by activated dendritic cells. J. Immunol. 2010, 184, 1876–1884. [Google Scholar] [CrossRef] [PubMed]
- Witalison, E.E.; Cui, X.; Hofseth, A.B.; Subramanian, V.; Causey, C.P.; Thompson, P.R.; Hofseth, L.J. Inhibiting protein arginine deiminases has antioxidant consequences. J. Pharmacol. Exp. Ther. 2015, 353, 64–70. [Google Scholar] [CrossRef] [PubMed]







© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jang, B.; Ishigami, A.; Kim, Y.-S.; Choi, E.-K. The Peptidylarginine Deiminase Inhibitor Cl-Amidine Suppresses Inducible Nitric Oxide Synthase Expression in Dendritic Cells. Int. J. Mol. Sci. 2017, 18, 2258. https://doi.org/10.3390/ijms18112258
Jang B, Ishigami A, Kim Y-S, Choi E-K. The Peptidylarginine Deiminase Inhibitor Cl-Amidine Suppresses Inducible Nitric Oxide Synthase Expression in Dendritic Cells. International Journal of Molecular Sciences. 2017; 18(11):2258. https://doi.org/10.3390/ijms18112258
Chicago/Turabian StyleJang, Byungki, Akihito Ishigami, Yong-Sun Kim, and Eun-Kyoung Choi. 2017. "The Peptidylarginine Deiminase Inhibitor Cl-Amidine Suppresses Inducible Nitric Oxide Synthase Expression in Dendritic Cells" International Journal of Molecular Sciences 18, no. 11: 2258. https://doi.org/10.3390/ijms18112258
APA StyleJang, B., Ishigami, A., Kim, Y. -S., & Choi, E. -K. (2017). The Peptidylarginine Deiminase Inhibitor Cl-Amidine Suppresses Inducible Nitric Oxide Synthase Expression in Dendritic Cells. International Journal of Molecular Sciences, 18(11), 2258. https://doi.org/10.3390/ijms18112258