Metabolic Dysfunction and Oxidative Stress in Epilepsy

Abstract
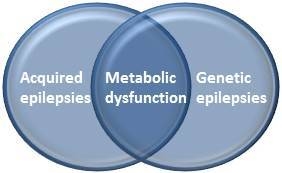
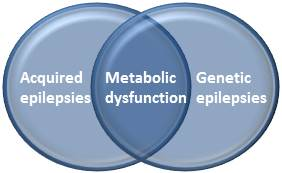
:
1. Introduction
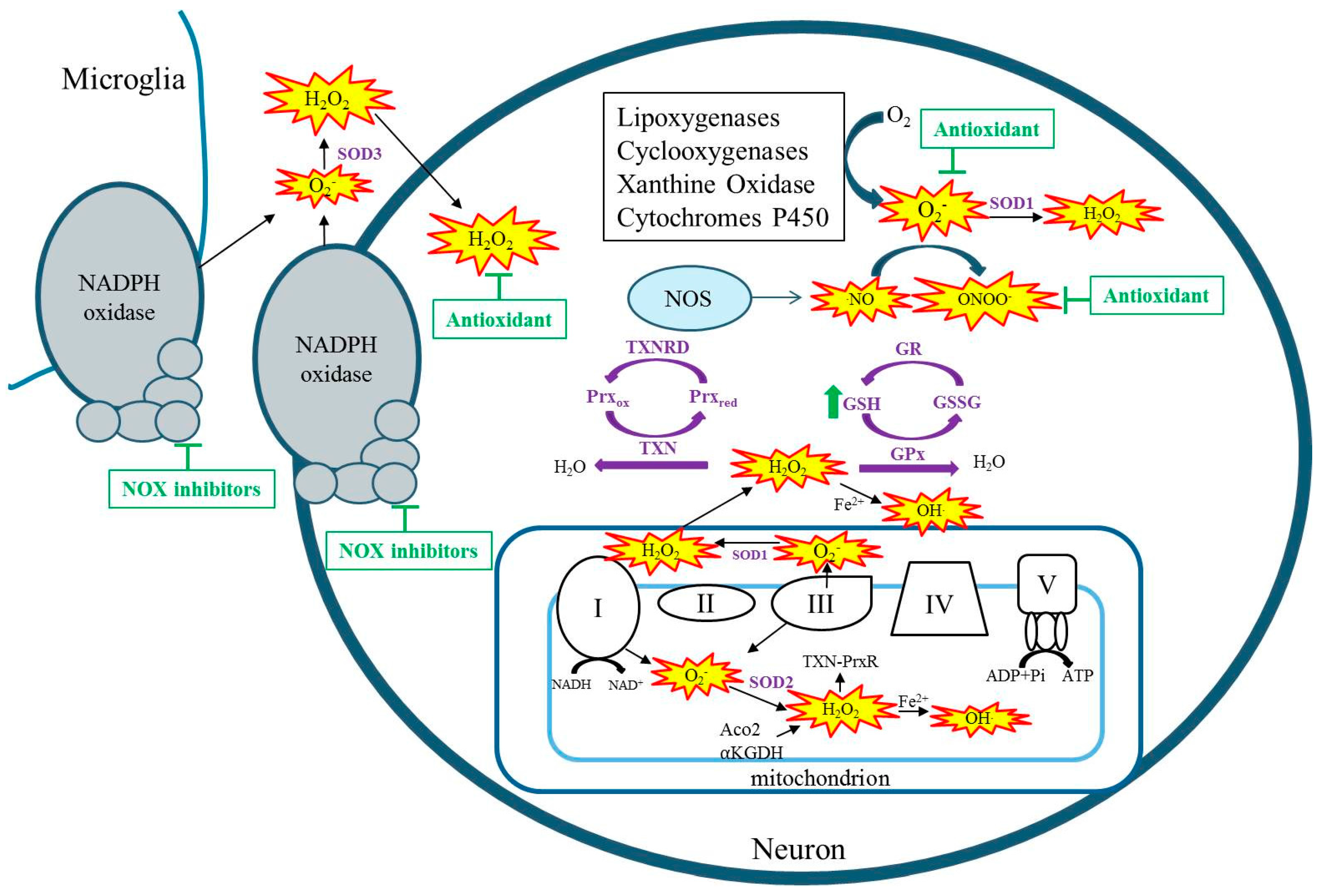
2. Sources of Reactive Species and Oxidative Stress
3. Oxidative Stress in Genetic Epilepsies
3.1. Mitochondrial Encephalopathies
3.2. Genetic Epilepsies Associated with Metabolic Dysfunction
3.3. Genetic Epilepsies and Antioxidant Systems
4. Oxidative Stress in Acquired Epilepsies
5. Oxidative Stress as a Therapeutic Target
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Institute of Medicine. Epilepsy across the Spectrum: Promoting Health and Understanding; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Porter, R.J.; Dhir, A.; Macdonald, R.L.; Rogawski, M.A. Chapter 39—Mechanisms of action of antiseizure drugs. In Handbook of Clinical Neurology; Hermann, S., William, H.T., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; Volume 108, pp. 663–681. [Google Scholar]
- Helmstaedter, C.; Kurthen, M.; Lux, S.; Reuber, M.; Elger, C.E. Chronic epilepsy and cognition: A longitudinal study in temporal lobe epilepsy. Ann. Neurol. 2003, 54, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Boveris, A. Determination of the production of superoxide radicals and hydrogen peroxide in mitochondria. Methods Enzymol. 1984, 105, 429–435. [Google Scholar] [PubMed]
- Gardner, P.R.; Fridovich, I. Inactivation-reactivation of aconitase in Escherichia coli. A sensitive measure of superoxide radical. J. Biol. Chem. 1992, 267, 8757–8763. [Google Scholar] [PubMed]
- Flint, D.H.; Tuminello, J.F.; Emptage, M.H. The inactivation of Fe-S cluster containing hydro-lyases by superoxide. J. Biol. Chem. 1993, 268, 22369–22376. [Google Scholar] [PubMed]
- Liang, L.P.; Ho, Y.S.; Patel, M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience 2000, 101, 563–570. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Liang, L.-P.; Hellier, J.L.; Staley, K.J.; Patel, M. Mitochondrial DNA damage and impaired base excision repair during epileptogenesis. Neurobiol. Dis. 2008, 30, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Cock, H.R.; Tong, X.; Hargreaves, I.P.; Heales, S.J.R.; Clark, J.B.; Patsalos, P.N.; Thom, M.; Groves, M.; Schapira, A.H.V.; Shorvon, S.D.; et al. Mitochondrial dysfunction associated with neuronal death following status epilepticus in rat. Epilepsy Res. 2002, 48, 157–168. [Google Scholar] [CrossRef]
- Kunz, W.S.; Kudin, A.P.; Vielhaber, S.; Blümcke, I.; Zuschratter, W.; Schramm, J.; Beck, H.; Elger, C.E. Mitochondrial complex I deficiency in the epileptic focus of patients with temporal lobe epilepsy. Ann. Neurol. 2000, 48, 766–773. [Google Scholar] [CrossRef]
- Ryan, K.; Backos, D.S.; Reigan, P.; Patel, M. Post-translational oxidative modification and inactivation of mitochondrial complex I in epileptogenesis. J. Neurosci. 2012, 32, 11250–11258. [Google Scholar] [CrossRef] [PubMed]
- Folbergrová, J.; Ješina, P.; Drahota, Z.; Lisý, V.; Haugvicová, R.; Vojtíšková, A.; Houštěk, J. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Exp. Neurol. 2007, 204, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Folbergrová, J.; Ješina, P.; Haugvicová, R.; Lisý, V.; Houštěk, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Tretter, L.; Adam-Vizi, V. Generation of reactive oxygen species in the reaction catalyzed by α-Ketoglutarate dehydrogenase. J. Neurosci. 2004, 24, 7771–7778. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.-B.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cantu, D.; Schaack, J.; Patel, M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS ONE 2009, 4, e7095. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Quantitative aspects of the production of superoxide anion radical by milk xanthine oxidase. J. Biol. Chem. 1970, 245, 4053–4057. [Google Scholar] [PubMed]
- David, W.; Infanger, R.V.S.; Davisson, R.L. NADPH Oxidases of the brain: Distribution, regulation, and function. Antioxid. Redox Signal. 2006, 8, 1583–1596. [Google Scholar]
- Kovac, S.; Domijan, A.M.; Walker, M.C.; Abramov, A.Y. Seizure activity results in calcium- and mitochondria-independent ROS production via NADPH and xanthine oxidase activation. Cell Death Dis. 2014, 5, e1442. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Jang, B.G.; Choi, B.Y.; Kim, H.S.; Sohn, M.; Chung, T.N.; Choi, H.C.; Song, H.K.; Suh, S.W. Post-treatment of an NADPH oxidase inhibitor prevents seizure-induced neuronal death. Brain Res. 2013, 1499, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Li, Q.-Y.; Chang, L.-Y.; Crapo, J.; Liang, L.-P. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J. Neurochem. 2005, 92, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Pestana, R.R.F.; Kinjo, E.R.; Hernandes, M.S.; Britto, L.R.G. Reactive oxygen species generated by NADPH oxidase are involved in neurodegeneration in the pilocarpine model of temporal lobe epilepsy. Neurosci. Lett. 2010, 484, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Stadtman, E.R. Protein oxidation in aging and age-related diseases. Ann. N. Y. Acad. Sci. 2001, 928, 22–38. [Google Scholar] [CrossRef] [PubMed]
- Wong-ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.M.; Tieleman, D.P.; Monticelli, L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys. J. 2007, 93, 4225–4236. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Uttara, B.; Singh, A.V.; Zamboni, P.; Mahajan, R.T. Oxidative stress and neurodegenerative diseases: A review of upstream and downstream antioxidant therapeutic options. Curr. Neuropharmacol. 2009, 7, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, G.; Moraes, C.T.; Shanske, S.; Oh, S.J.; DiMauro, S. A new mtDNA mutation in the tRNA(Lys) gene associated with myoclonic epilepsy and ragged-red fibers (MERRF). Am. J. Hum. Genet. 1992, 51, 1213–1217. [Google Scholar] [PubMed]
- Wu, S.-B.; Ma, Y.-S.; Wu, Y.-T.; Chen, Y.-C.; Wei, Y.-H. Mitochondrial DNA mutation-elicited oxidative stress, oxidative damage, and altered gene expression in cultured cells of patients with MERRF syndrome. Mol. Neurobiol. 2010, 41, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Pinton, P.; King, M.P.; Davidson, M.; Schon, E.A.; Rizzuto, R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 1999, 5, 951–954. [Google Scholar] [PubMed]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar] [PubMed]
- Quintana, A.; Kruse, S.E.; Kapur, R.P.; Sanz, E.; Palmiter, R.D. Complex I deficiency due to loss of Ndufs4 in the brain results in progressive encephalopathy resembling Leigh syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 10996–11001. [Google Scholar] [CrossRef] [PubMed]
- Wojtala, A.; Karkucinska-Wieckowska, A.; Sardao, V.A.; Szczepanowska, J.; Kowalski, P.; Pronicki, M.; Duszynski, J.; Wieckowski, M.R. Modulation of mitochondrial dysfunction-related oxidative stress in fibroblasts of patients with Leigh syndrome by inhibition of prooxidative p66Shc pathway. Mitochondrion 2017. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR inhibition alleviates mitochondrial disease in a mouse model of leigh syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Naviaux, R.K.; Nyhan, W.L.; Barshop, B.A.; Poulton, J.; Markusic, D.; Karpinski, N.C.; Haas, R.H. Mitochondrial DNA polymerase γ deficiency and mtDNA depletion in a child with Alpers’ syndrome. Ann. Neurol. 1999, 45, 54–58. [Google Scholar] [CrossRef]
- Saneto, R.P. Alpers-Huttenlocher syndrome: The role of a multidisciplinary health care team. J. Multidiscip. Healthc. 2016, 9, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Simonati, A.; Filosto, M.; Savio, C.; Tomelleri, G.; Tonin, P.; Dalla Bernardina, B.; Rizzuto, N. Features of cell death in brain and liver, the target tissues of progressive neuronal degeneration of childhood with liver disease (Alpers-Huttenlocher disease). Acta Neuropathol. 2003, 106, 57–65. [Google Scholar] [PubMed]
- Morris, A.A.M.; Singh-Kler, R.; Perry, R.H.; Griffiths, P.D.; Burt, A.D.; Wong, C.P.; Gardner-Medwin, D.; Tumbull, D.M. Respiratory chain dysfunction in progressive neuronal degeneration of childhood with liver disease. J. Child Neurol. 1996, 11, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Jamme, I.; Petit, E.; Divoux, D.; Alain, G.; Maixent, J.M.; Nouvelot, A. Modulation of mouse cerebral Na+, K+-ATPase activity by oxygen free radicals. Neuroreport 1995, 7, 333–337. [Google Scholar] [PubMed]
- Fighera, M.R.; Royes, L.F.F.; Furian, A.F.; Oliveira, M.S.; Fiorenza, N.G.; Frussa-Filho, R.; Petry, J.C.; Coelho, R.C.; Mello, C.F. GM1 ganglioside prevents seizures, Na+,K+-ATPase activity inhibition and oxidative stress induced by glutaric acid and pentylenetetrazole. Neurobiol. Dis. 2006, 22, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; Trouillard, O.; Saint-Martin, C.; Gourfinkel-An, I.; Bouteiller, D.; Carpentier, W.; Keren, B.; Abert, B.; Gautier, A.; Baulac, S.; et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: Analysis of 333 patients. J. Med. Genet. 2009, 46, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kinsman, S.L.; Vining, E.P.G.; Quaskey, S.A.; Mellits, D.; Freeman, J.M. Efficacy of the ketogenic diet for intractable seizure disorders: Review of 58 cases. Epilepsia 1992, 33, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Veggiotti, P.; Burlina, A.; Coppola, G.; Cusmai, R.; De Giorgis, V.; Guerrini, R.; Tagliabue, A.; Bernardina, B.D. The ketogenic diet for Dravet syndrome and other epileptic encephalopathies: An Italian consensus. Epilepsia 2011, 52, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Wirrell, E.C.; Laux, L.; Franz, D.N.; Sullivan, J.; Saneto, R.P.; Morse, R.P.; Devinsky, O.; Chugani, H.; Hernandez, A.; Hamiwka, L.; et al. Stiripentol in dravet syndrome: Results of a retrospective U.S. study. Epilepsia 2013, 54, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Chiron, C.; Marchand, M.C.; Tran, A.; Rey, E.; d’Athis, P.; Vincent, J.; Dulac, O.; Pons, G. Stiripentol in severe myoclonic epilepsy in infancy: A randomised placebo-controlled syndrome-dedicated trial. Lancet 2000, 356, 1638–1642. [Google Scholar] [CrossRef]
- Inoue, Y.; Ohtsuka, Y. Long-term safety and efficacy of stiripentol for the treatment of Dravet syndrome: A multicenter, open-label study in Japan. Epilepsy Res. 2015, 113, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Doccini, S.; Meschini, M.C.; Mei, D.; Guerrini, R.; Sicca, F.; Santorelli, F.M. Mitochondrial respiratory chain defects in skin fibroblasts from patients with Dravet syndrome. Neurol. Sci. 2015, 36, 2151–2155. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.G.; Rowley, S.; Fulton, R.; Dinday, M.T.; Baraban, S.C.; Patel, M. Altered Glycolysis and Mitochondrial Respiration in a zebrafish model of Dravet syndrome. eNeuro 2016, 3. [Google Scholar] [CrossRef] [PubMed]
- Baraban, S.C.; Dinday, M.T.; Hortopan, G.A. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat. Commun. 2013, 4, 2410. [Google Scholar] [CrossRef] [PubMed]
- Fehm, H.L.; Kern, W.; Peters, A. The selfish brain: Competition for energy resources. In Progress in Brain Research; Kalsbeek, A., Fliers, E., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 153, pp. 129–140. [Google Scholar]
- De Lores Arnaiz, G.R.; Ordieres, M.G.L. Brain Na(+), K(+)-ATPase activity in aging and disease. Int. J. Biomed. Sci. 2014, 10, 85–102. [Google Scholar] [PubMed]
- Klepper, J.; Diefenbach, S.; Kohlschütter, A.; Voit, T. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot. Essent. Fatty Acids 2004, 70, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Okumura, A.; Hayashi, M.; Mori, H.; Takahashi, S.; Yanagihara, K.; Miyata, R.; Tanuma, N.; Mimaki, T.; Abe, S.; et al. Oxidative stress markers and phosphorus magnetic resonance spectroscopy in a patient with GLUT1 deficiency treated with modified Atkins diet. Brain Dev. 2012, 34, 372–375. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Ko, K.H.; Kim, W.-K.; Chae, J.S.; Yen, T.P.H.; Kim, H.J.; Wie, M.-B.; Kim, H.-C. Role of glutathione peroxidase in the ontogeny of hippocampal oxidative stress and kainate seizure sensitivity in the genetically epilepsy-prone rats. Neurochem. Int. 2008, 52, 1134–1147. [Google Scholar] [CrossRef] [PubMed]
- Melov, S.; Coskun, P.; Patel, M.; Tuinstra, R.; Cottrell, B.; Jun, A.S.; Zastawny, T.H.; Dizdaroglu, M.; Goodman, S.I.; Huang, T.-T.; et al. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc. Natl. Acad. Sci. USA 1999, 96, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-P.; Waldbaum, S.; Rowley, S.; Huang, T.-T.; Day, B.J.; Patel, M. Mitochondrial oxidative stress and epilepsy in SOD2 deficient mice: Attenuation by a lipophilic metalloporphyrin. Neurobiol. Dis. 2012, 45, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.-P.; Patel, M. Mitochondrial oxidative stress and increased seizure susceptibility in Sod2−/+ mice. Free Radic. Biol. Med. 2004, 36, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Danbolt, N.C.; Volterra, A. Glutamate transporters are oxidant-vulnerable: A molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998, 19, 328–334. [Google Scholar] [CrossRef]
- Carmel-Harel, O.; Storz, G. Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and saccharomyces cerevisiae responses to oxidative stress. Annu. Rev. Microbiol. 2000, 54, 439–461. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 Removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Baron, G.; Zsurka, G.; Hampel, K.G.; Elger, C.E.; Grote, A.; Weber, Y.; Lerche, H.; Thiele, H.; Nürnberg, P.; et al. Homozygous mutation in TXNRD1 is associated with genetic generalized epilepsy. Free Radic. Biol. Med. 2017, 106, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; Kobayashi, S.; Terrile, C.; Wolf, P.; Schaper, J.; et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain 2016, 139, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Takagi, Y.; Hattori, I.; Nozaki, K.; Mitsui, A.; Ishikawa, M.; Hashimoto, N.; Yodoi, J. Excitotoxic hippocampal injury is attenuated in thioredoxin transgenic mice. J. Cereb. Blood Flow Metab. 2000, 20, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Mahyar, A.; Ayazi, P.; Fallahi, M.; Javadi, A. Correlation between serum selenium level and febrile seizures. Pediatr. Neurol. 2010, 43, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.R.; Shams, S.; Nouri, M.; Mohseni, M.; Shabanian, R.; Yekaninejad, M.S.; Chegini, N.; Khodadad, A.; Safaralizadeh, R. A Probable causative factor for an old problem: Selenium and glutathione peroxidase appear to play important roles in epilepsy pathogenesis. Epilepsia 2007, 48, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, M.R.; Shabanian, R.; Abbaskhanian, A.; Nasirian, A.; Ghofrani, M.; Mohammadi, M.; Zamani, G.R.; Kayhanidoost, Z.; Ebrahimi, S.; Pourpak, Z. Selenium and intractable epilepsy: Is there any correlation? Pediatr. Neurol. 2007, 36, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Seven, M.; Basaran, S.Y.; Cengiz, M.; Unal, S.; Yuksel, A. Deficiency of selenium and zinc as a causative factor for idiopathic intractable epilepsy. Epilepsy Res. 2013, 104, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Kutluhan, S.; Yılmaz, M. Selenium and topiramate modulates brain microsomal oxidative stress values, Ca2+-ATPase activity, and EEG records in pentylentetrazol-induced seizures in rats. J. Membr. Biol. 2008, 225, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Rehni, A.K.; Singh, T.G. Selenium induced anticonvulsant effect: A potential role of prostaglandin E1 receptor activation linked mechanism. J. Trace Elem. Med. Biol. 2013, 27, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int. J. Stroke 2009, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Ana, R.-R.; Juan Jose, E.-G.; Francisco, M.-C.; Antonio, C.-V. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar]
- Puttachary, S.; Sharma, S.; Stark, S.; Thippeswamy, T. Seizure-induced oxidative stress in temporal lobe epilepsy. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Kovac, S.; Dinkova Kostova, A.T.; Herrmann, A.M.; Melzer, N.; Meuth, S.G.; Gorji, A. Metabolic and homeostatic changes in seizures and acquired epilepsy—mitochondria, calcium dynamics and reactive oxygen species. Int. J. Mol. Sci. 2017, 18, 1935. [Google Scholar] [CrossRef] [PubMed]
- Ristić, A.J.; Savić, D.; Sokić, D.; Bogdanović Pristov, J.; Nestorov, J.; Baščarević, V.; Raičević, S.; Savić, S.; Spasojević, I. Hippocampal antioxidative system in mesial temporal lobe epilepsy. Epilepsia 2015, 56, 789–799. [Google Scholar] [CrossRef] [PubMed]
- López, J.; González, M.E.; Lorigados, L.; Morales, L.; Riverón, G.; Bauzá, J.Y. Oxidative stress markers in surgically treated patients with refractory epilepsy. Clin. Biochem. 2007, 40, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Yüzbaşioğlu, A.; Karataş, H.; Gürsoy-Özdemir, Y.; Saygi, S.; Akalan, N.; Söylemezoğlu, F.; Dalkara, T.; Kocaefe, Y.Ç.; Özgüç, M. Changes in the expression of selenoproteins in mesial temporal lobe epilepsy patients. Cell. Mol. Neurobiol. 2009, 29, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Pecorelli, A.; Natrella, F.; Belmonte, G.; Miracco, C.; Cervellati, F.; Ciccoli, L.; Mariottini, A.; Rocchi, R.; Vatti, G.; Bua, A.; et al. NADPH oxidase activation and 4-hydroxy-2-nonenal/aquaporin-4 adducts as possible new players in oxidative neuronal damage presents in drug-resistant epilepsy. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 507–519. [Google Scholar] [CrossRef] [PubMed]
- Rumià, J.; Marmol, F.; Sanchez, J.; Giménez-Crouseilles, J.; Carreño, M.; Bargalló, N.; Boget, T.; Pintor, L.; Setoain, X.; Donaire, A.; et al. Oxidative stress markers in the neocortex of drug-resistant epilepsy patients submitted to epilepsy surgery. Epilepsy Res. 2013, 107, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Liang, L.-P.; Roberts Ii, L.J. Enhanced hippocampal F2-isoprostane formation following kainate-induced seizures. J. Neurochem. 2001, 79, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.N.; Warren, E.; Liang, L.-P.; Roberts, L.J.; Patel, M. Scavenging of highly reactive gamma-ketoaldehydes attenuates cognitive dysfunction associated with epileptogenesis. Neurobiol. Dis. 2017, 98, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, A.; Kawamoto, Y.; Chiba, Y.; Takei, S.; Hasegawa-Ishii, S.; Kawamura, N.; Yoshikawa, K.; Hosokawa, M.; Oikawa, S.; Kato, M.; et al. Proteomic identification of hippocampal proteins vulnerable to oxidative stress in excitotoxin-induced acute neuronal injury. Neurobiol. Dis. 2011, 43, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Waldbaum, S.; Patel, M. Mitochondrial oxidative stress in temporal lobe epilepsy. Epilepsy Res. 2010, 88, 23–45. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Henshall, D.C. Apoptosis signalling pathways in seizure-induced neuronal death and epilepsy. Biochem. Soc. Trans. 2007, 35, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Niquet, J.; Liu, H.; Wasterlain, C.G. Programmed neuronal necrosis and status epilepticus. Epilepsia 2005, 46, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Henshall, D.C.; Engel, T. Contribution of apoptosis-associated signaling pathways to epileptogenesis: Lessons from Bcl-2 family knockouts. Front. Cell. Neurosci. 2013, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, I.; Hastings, T. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J. Neurosci. 1995, 15, 3318–3327. [Google Scholar] [PubMed]
- Henshall, D.C.; Murphy, B.M. Modulators of neuronal cell death in epilepsy. Curr. Opin. Pharm. 2008, 8, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef]
- Lafon-Cazal, M.; Pietri, S.; Culcasi, M.; Bockaert, J. NMDA-dependent superoxide production and neurotoxicity. Nature 1993, 364, 535–537. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Blomgren, K.; Kroemer, G. Mitochondrial membrane permeabilization in neuronal injury. Nat. Rev. Neurosci. 2009, 10, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.G.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, R.S.; Strasser, A.; McDunn, J.E.; Swanson, P.E. Cell death in disease: Mechanisms and emerging therapeutic concepts. N. Engl. J. Med. 2009, 361, 1570–1583. [Google Scholar] [CrossRef] [PubMed]
- Frantseva, M.V.; Perez Velazquez, J.L.; Tsoraklidis, G.; Mendonca, A.J.; Adamchik, Y.; Mills, L.R.; Carlen, P.L.; Burnham, M.W. Oxidative stress is involved in seizure-induced neurodegeneration in the kindling model of epilepsy. Neuroscience 2000, 97, 431–435. [Google Scholar] [CrossRef]
- Rong, Y.; Doctrow, S.R.; Tocco, G.; Baudry, M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc. Natl. Acad. Sci. USA 1999, 96, 9897–9902. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.-Y.; Han, S.-H. Melatonin attenuates kainic acid-induced hippocampal neurodegeneration and oxidative stress through microglial inhibition. J. Pineal Res. 2003, 34, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.-X.; Manchester, L.C.; Reiter, R.J.; Qi, W.; Kim, S.J.; El-Sokkary, G.H. Melatonin protects hippocampal neurons in vivo against kainic acid-induced damage in mice. J. Neurosci. Res. 1998, 54, 382–389. [Google Scholar] [CrossRef]
- MacGregor, D.G.; Higgins, M.J.; Jones, P.A.; Maxwell, W.L.; Watson, M.W.; Graham, D.I.; Stone, T.W. Ascorbate attenuates the systemic kainate-induced neurotoxicity in the rat hippocampus. Brain Res. 1996, 727, 133–144. [Google Scholar] [CrossRef]
- Pauletti, A.; Terrone, G.; Shekh-Ahmad, T.; Salamone, A.; Ravizza, T.; Rizzi, M.; Pastore, A.; Pascente, R.; Liang, L.-P.; Villa, B.R.; et al. Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 2017, 140, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.N.; Rowley, S.; Liang, L.-P.; White, A.M.; Day, B.J.; Patel, M. Reactive oxygen species mediate cognitive deficits in experimental temporal lobe epilepsy. Neurobiol. Dis. 2015, 82, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Sutula, T.P. Is epilepsy a progressive disorder? Prospects for new therapeutic approaches in temporal-lobe epilepsy. Lancet Neurol. 2002, 1, 173–181. [Google Scholar] [CrossRef]
- Barros, D.O.; Xavier, S.M.L.; Barbosa, C.O.; Silva, R.F.; Freitas, R.L.M.; Maia, F.D.; Oliveira, A.A.; Freitas, R.M.; Takahashi, R.N. Effects of the vitamin E in catalase activities in hippocampus after status epilepticus induced by pilocarpine in Wistar rats. Neurosci. Lett. 2007, 416, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.L.; Burnham, W.M.; Hwang, P.A. An evaluation of the anticonvulsant effects of vitamin E. Epilepsy Res. 1990, 6, 12–17. [Google Scholar] [CrossRef]
- Ogunmekan, A.O.; Hwang, P.A. A Randomized, Double-Blind, Placebo-Controlled, Clinical Trial of D-α-Tocopheryl Acetate (Vitamin E), as Add-On Therapy, for Epilepsy in Children. Epilepsia 1989, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Mehvari, J.; Motlagh, F.G.; Najafi, M.; Ghazvini, M.R.A.; Naeini, A.A.; Zare, M. Effects of Vitamin E on seizure frequency, electroencephalogram findings, and oxidative stress status of refractory epileptic patients. Adv. Biomed. Res. 2016, 5, 36. [Google Scholar] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Disorder | Gene Mutation | Resulting Dysfuntion | Finding | Citation |
|---|---|---|---|---|
| MERFF | tRNALys | Complex I | Decreased ATP, increased ROS, altered antioxidant gene expression, alterations to calcium homeostatsis | [32,33,34] |
| Leigh syndrome | Various mtDNA mutations | Complex I, V | Increased ROS, decreased ATP | [35,36,37] |
| AHS | POLG | Decreased mtDNA, Complex IV | Increased apoptosis and necrosis potentially modulated by mito pathways | [40,42] |
| Dravet Syndrome | SCN1A | Nav1.1 | In zebrafish—decreased glycolytic and oxygen consumption rates, downregulation of glycolytic pathway | [53] |
| Glut1 deficiency | SLC2A1 | Glucose transport into brain | Increased oxidative DNA damage, increased lipid peroxidation—attenuated by modified Atkins diet | [58] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearson-Smith, J.N.; Patel, M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. Int. J. Mol. Sci. 2017, 18, 2365. https://doi.org/10.3390/ijms18112365
Pearson-Smith JN, Patel M. Metabolic Dysfunction and Oxidative Stress in Epilepsy. International Journal of Molecular Sciences. 2017; 18(11):2365. https://doi.org/10.3390/ijms18112365
Chicago/Turabian StylePearson-Smith, Jennifer N., and Manisha Patel. 2017. "Metabolic Dysfunction and Oxidative Stress in Epilepsy" International Journal of Molecular Sciences 18, no. 11: 2365. https://doi.org/10.3390/ijms18112365
APA StylePearson-Smith, J. N., & Patel, M. (2017). Metabolic Dysfunction and Oxidative Stress in Epilepsy. International Journal of Molecular Sciences, 18(11), 2365. https://doi.org/10.3390/ijms18112365