Lp16-PSP, a Member of YjgF/YER057c/UK114 Protein Family Induces Apoptosis and p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in Human Acute Promyelocytic Leukemia (APL) HL-60 Cells

Abstract
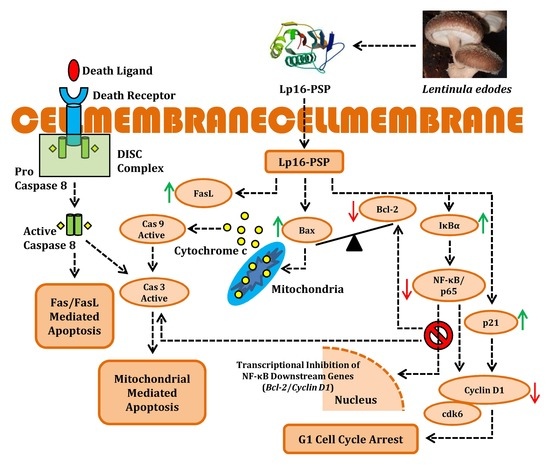
:
1. Introduction
2. Results
2.1. Cytotoxic Activity of Lp16-PSP in Human Acute Promyeloid Leukemia (HL-60 Cells)
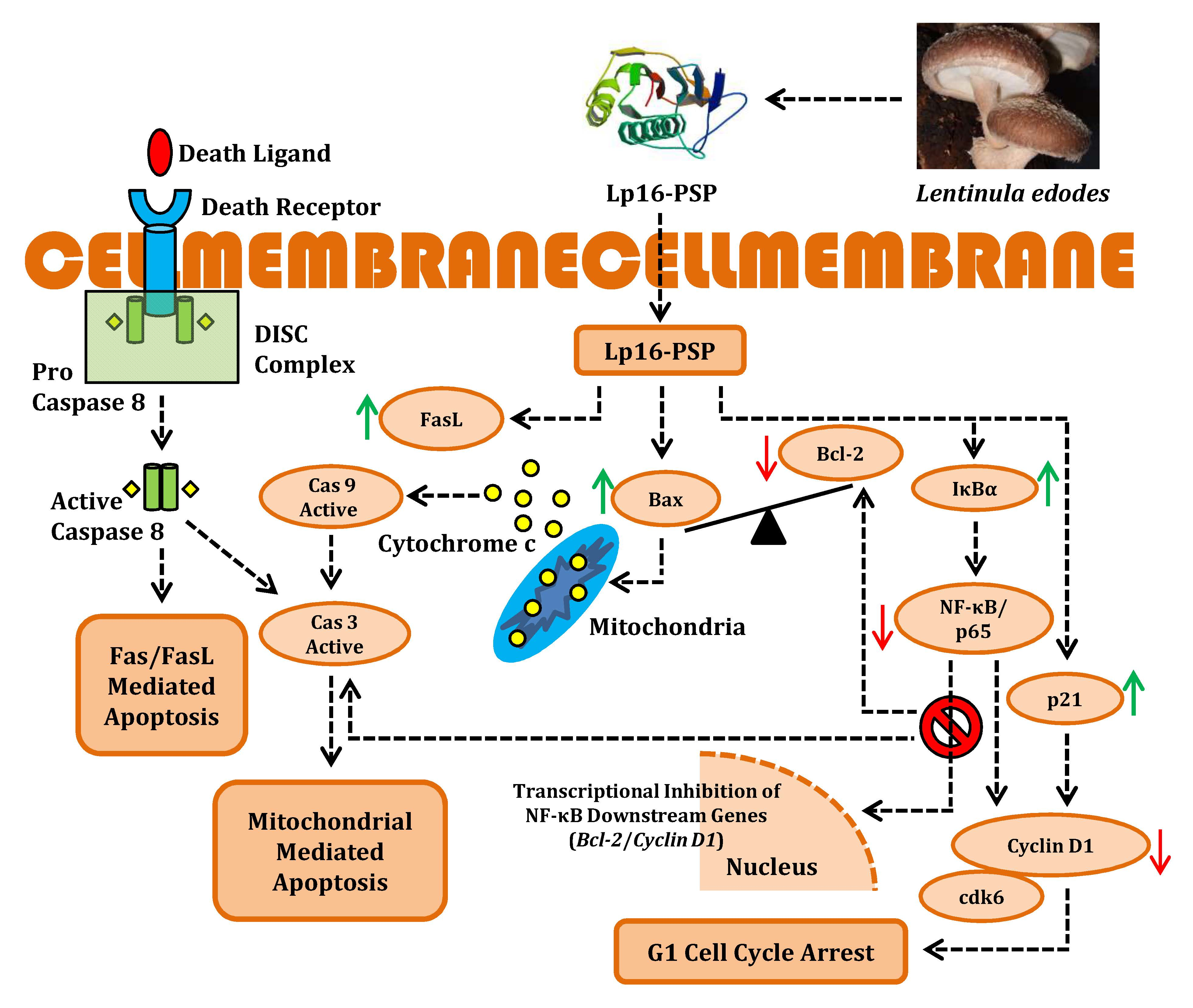
2.2. Lp16-PSP-Induced Nuclear Morphological Change and DNA Fragmentation in Acute Promyeloid Leukemia (APL) HL-60 Cells
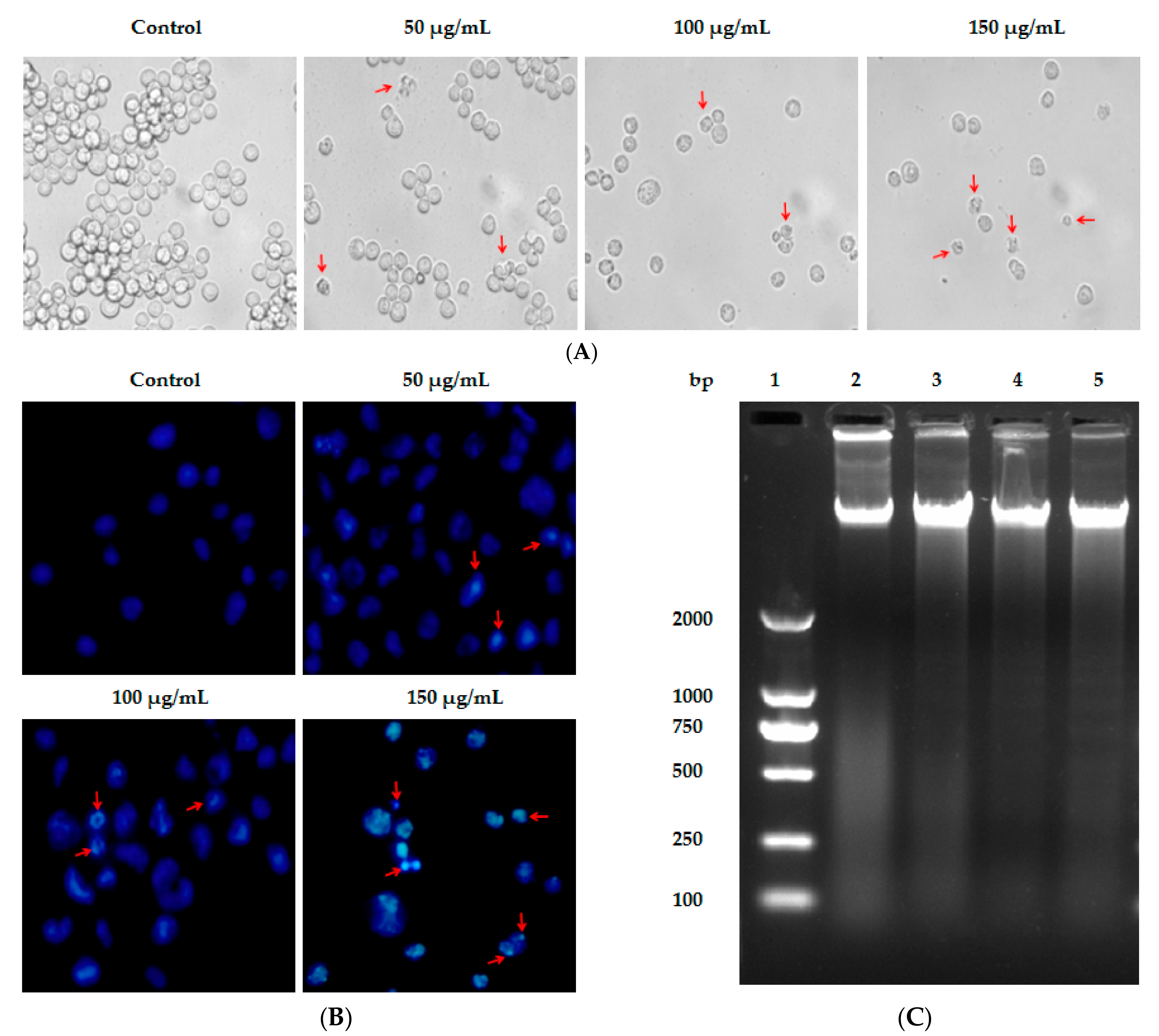
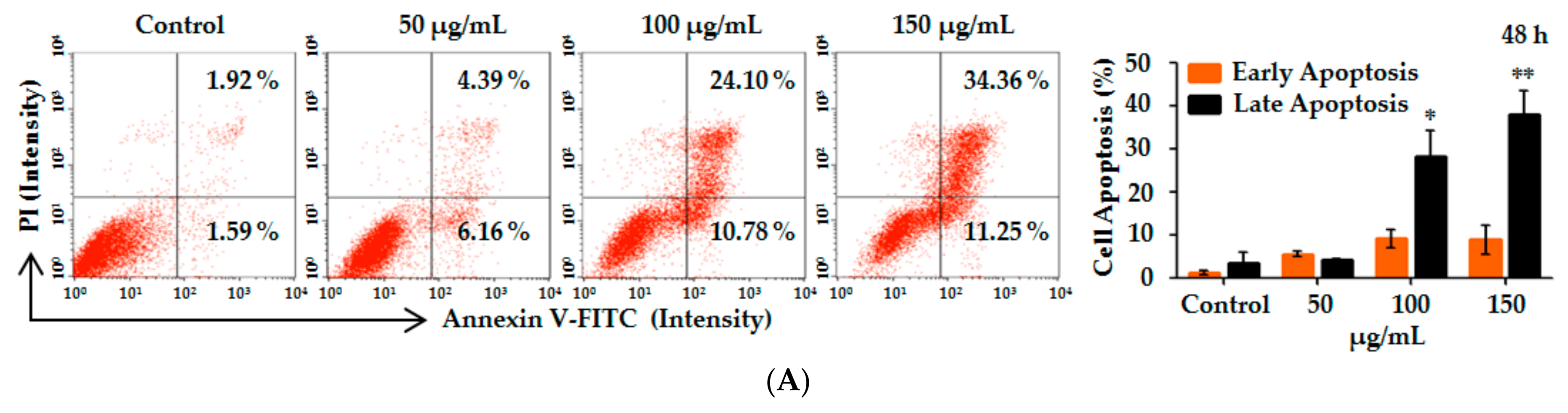
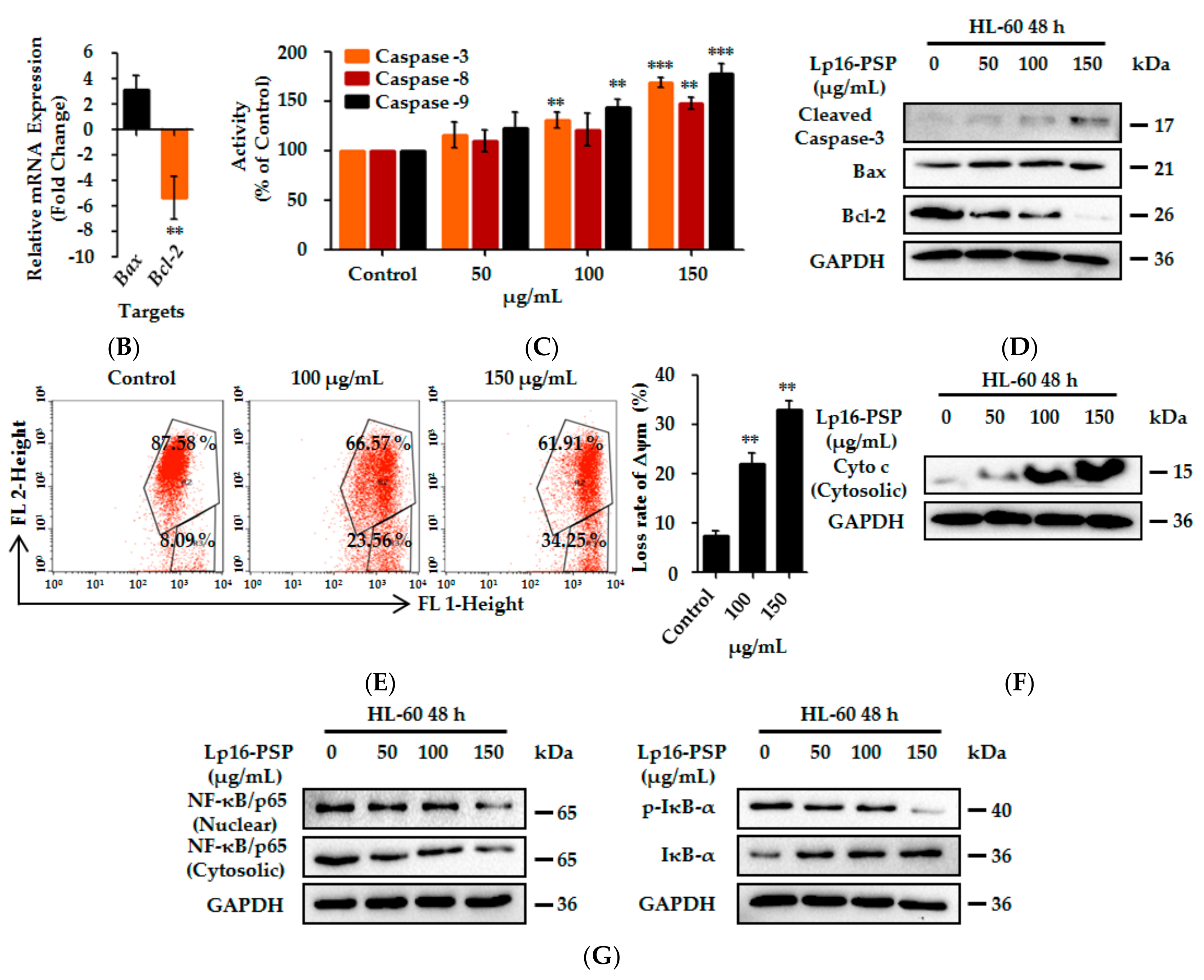
2.3. Lp16-PSP-Induced Apoptosis and Involvement of Extrinsic and Intrinsic Pathways
2.4. Lp16-PSP Inhibits the Consecutive Translocation of NF-κB/p65 into the Nucleus in HL-60 Cells
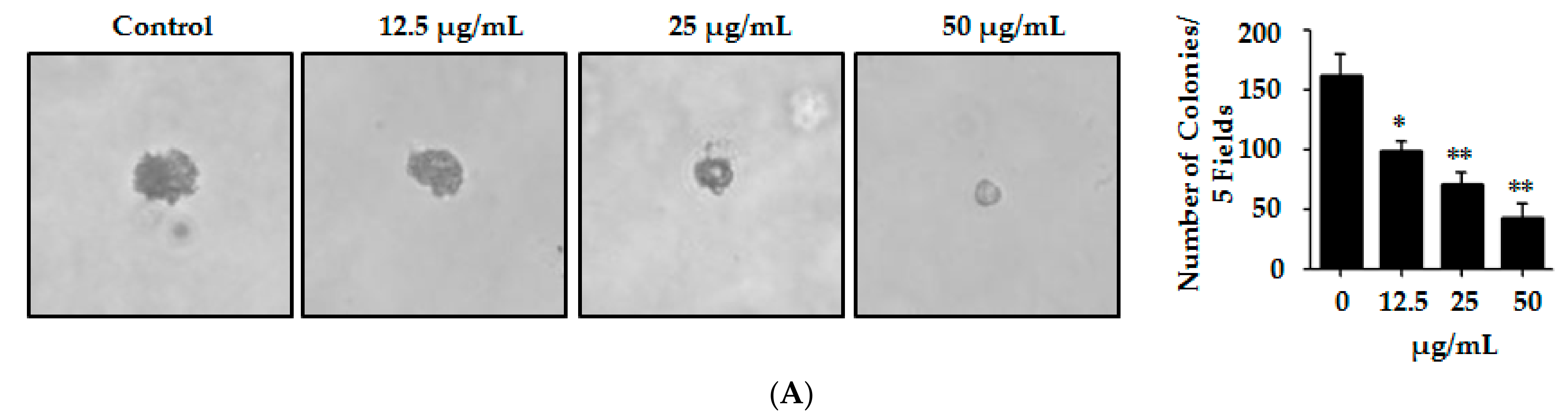
2.5. Lp16-PSP Suppresses the Anchorage-Independent Colony Formation of HL-60 Cells
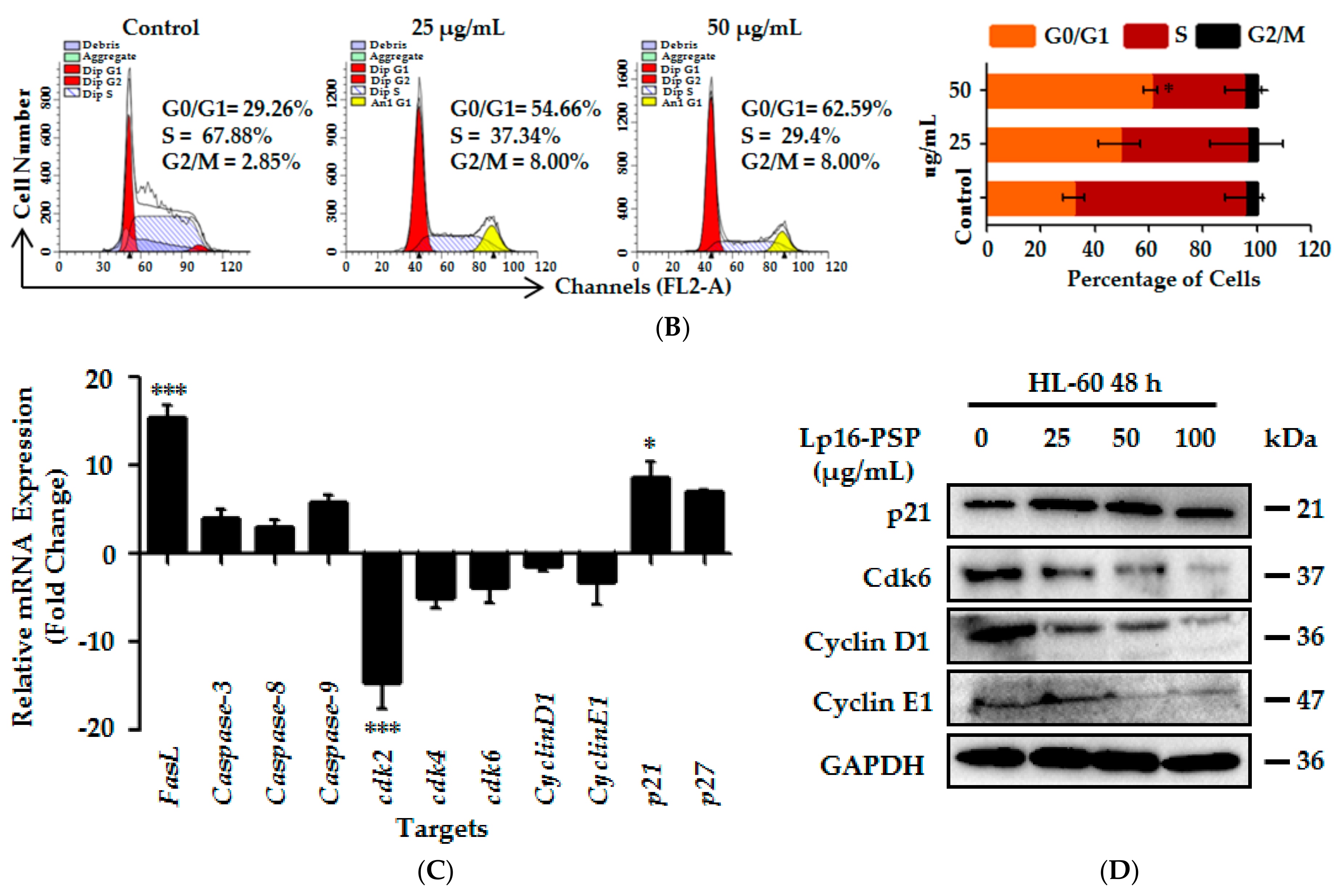
2.6. Lp16-PSP Induces G1 Phase Cell Cycle Arrest in HL-60 Cells
2.7. Lp16-PSP Induced p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in HL-60
3. Discussion
4. Material and Methods
4.1. Expression of the Recombinant Protein Lp16-PSP
4.2. Human Leukemia HL-60 Cells and Culture Conditions
4.3. Antibodies, Kits, and Reagents
4.4. Phase Contrast Imaging
4.5. Hoechst 33258 Staining
4.6. DNA Fragmentation Assay
4.7. Colorimetric Analysis of Caspase-3, -8, and -9
4.8. Apoptosis Analysis Using Annexin-V-FITC/PI Staining
4.9. Mitochondrial Membrane Potential (Δψm) Measurement Using JC-1 Staining by Flow Cytometry
4.10. Soft-Agar Colony Formation Assay
4.11. Cell-Cycle Analysis by Flow Cytometry
4.12. Isolation of RNA and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.13. Western Blotting
4.14. Statistical Evaluation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, M.R.; Ha, P.J. Haematology: An Illustrated Colour Text, 2nd ed.; Churchill Livingstone: London, UK, 2002. [Google Scholar]
- Recher, C.; Beyne-Rauzy, O.; Demur, C.; Chicanne, G.; dos Santos, C.; Mas, V.M.; Benzaquen, D.; Laurent, G.; Huguet, F.; Payrastre, B. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood 2005, 105, 2527–2534. [Google Scholar] [CrossRef] [PubMed]
- Coombs, C.C.; Tavakkoli, M.; Tallman, M.S. Acute promyelocytic leukemia: Where did we start, where are we now, and the future. Blood Cancer J. 2015, 5, e304. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.A.; lo Coco, F.; Martin, G.; Avvisati, G.; Rayon, C.; Barbui, T.; Díaz-Mediavilla, J.; Fioritoni, G.; González, J.D.; Liso, V.; et al. Definition of relapse risk and role of nonanthracycline drugs for consolidation in patients with acute promyelocytic leukemia: A joint study of the PETHEMA and GIMEMA cooperative groups. Blood 2000, 96, 1247–1253. [Google Scholar] [PubMed]
- Tallman, M.S.; Andersen, J.W.; Schiffer, C.A.; Appelbaum, F.R.; Feusner, J.H.; Ogden, A.; Weinstein, H.; Shepherd, L.; Willman, C.; Bloomfield, C.D.; et al. All-trans-retinoic acid in acute promyelocytic leukemia. N. Engl. J. Med. 1997, 337, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A. Foye’s Principles of Medicinal Chemistry; Lemke, T., Williams, D.A., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Baltimore, MD, USA, 2008; pp. 12–25. [Google Scholar]
- Lucas, D.M.; Still, P.C.; Perez, L.B.; Grever, M.R.; Kinghorn, A.D. Potential of plant-derived natural products in the treatment of leukemia and lymphoma. Curr. Drug Targets 2010, 11, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Saedi, T.A.; Md Noor, S.; Ismail, P.; Othman, F. The Effects of Herbs and Fruits on Leukaemia. Evid. Based Complement. Altern. Med. 2014, 2014, 494136. [Google Scholar] [CrossRef] [PubMed]
- Gurova, K. New hopes from old drugs: Revisiting DNA-binding small molecules as anticancer agents. Future Oncol. Lond. Engl. 2009, 5, 1685–1704. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, G.; Ercole, C.; Lista, M.; Pizzo, E.; Falanga, A.; Galdiero, S.; Spadaccini, R.; Picone, D. Enforcing the positive charge of N-termini enhances membrane interaction and antitumor activity of bovine seminal ribonuclease. Biochim. Biophys. Acta 2011, 1808, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Zhang, C.Z.; Zhang, L.; Fong, W.P.; Ng, T.B. In vitro and in vivo anticarcinogenic effects of RNase MC2, a ribonuclease isolated from dietary bitter gourd, toward human liver cancer cells. Int. J. Biochem. Cell Biol. 2012, 44, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Zhang, C.Z.; Fong, W.P.; Ng, T.B. RNase MC2: A new Momordica charantia ribonuclease that induces apoptosis in breast cancer cells associated with activation of MAPKs and induction of caspase pathways. Apoptosis Int. J. Program. Cell Death 2012, 17, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Lomax, J.E.; Eller, C.H.; Raines, R.T. Rational design and evaluation of mammalian ribonuclease cytotoxins. Methods Enzymol. 2012, 502, 273–290. [Google Scholar] [PubMed]
- Joseph, T.P.; Chanda, W. Expression and selective in vitro anticancer activity of Lp16-PSP, a member of YjgF/YER057c/UK114 protein family from the mushroom Lentinula edodes C91-3. Int. J. Mol. Med. 2017. under review. [Google Scholar]
- Oka, T.; Tsuji, H.; Noda, C.; Sakai, K.; Hong, Y.M.; Suzuki, I.; Muñoz, S.; Natori, Y. Isolation and characterization of a novel perchloric acid-soluble protein inhibiting cell-free protein synthesis. J. Biol. Chem. 1995, 270, 30060–30067. [Google Scholar] [PubMed]
- Asagi, K.; Oka, T.; Arao, K.; Suzuki, I.; Thakur, M.K.; Izumi, K.; Natori, Y. Purification, characterization and differentiation-dependent expression of a perchloric acid soluble protein from rat kidney. Nephron 1998, 79, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Goupil-Feuillerat, N.; Cocaign-Bousquet, M.; Godon, J.J.; Ehrlich, S.D.; Renault, P. Dual role of α-acetolactate decarboxylase in Lactococcus lactis subsp. lactis. J. Bacteriol. 1997, 179, 6285–6293. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Kawagoshi, A.; Sawasaki, T.; Madin, K.; Ogasawara, T.; Oka, T.; Endo, Y. Ribonuclease activity of rat liver perchloric acid-soluble protein, a potent inhibitor of protein synthesis. J. Biol. Chem. 1999, 274, 20688–20692. [Google Scholar] [CrossRef] [PubMed]
- Manjasetty, B.A.; Delbruck, H.; Pham, D.T.; Mueller, U.; Fieber-Erdmann, M.; Scheich, C.; Sievert, V.; Büssow, K.; Niesen, F.H.; Weihofen, W.; et al. Crystal structure of Homo sapiens protein hp14.5. Proteins 2004, 54, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Ceciliani, F.; Faotto, L.; Negri, A.; Colombo, I.; Berra, B.; Bartorelli, A.; Ronchia, S. The primary structure of UK 114 tumor antigen. FEBS Lett. 1996, 393, 147–150. [Google Scholar] [CrossRef]
- Bartorelli, A.; Zanolo, G.; Fassio, F.; Botta, M.; Ferrara, R.; Bailo, M.; Cavalca, C.B.V.; Arzani, C.; Maraschin, R. Toxicological and antitumoral activity of UK101, a mammalian liver extract. J. Tumor Marker Oncol. 1994, 9, 49. [Google Scholar]
- Racca, S.; di Carlo, F.; Bartorelli, A.; Bussolati, B.; Bussolati, G. Growth inhibition of DMBA-induced rat mammary carcinomas by UK114 treatment. Virchows Arch. 1997, 431, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Ghezzo, F.; Berta, G.N.; Bussolati, B.; Bosio, A.; Corvetti, G.; di Carlo, F.; Bussolati, G.; Guglielmone, R.; Bartorelli, A. Perchloric acid-soluble proteins from goat liver inhibit chemical carcinogenesis of Syrian hamster cheek-pouch carcinoma. Br. J. Cancer 1999, 79, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.; Nardai, G.; Csermely, P.; Tompa, P.; Friedrich, P. DUK114, the Drosophila orthologue of bovine brain calpain activator protein, is a molecular chaperone. Biochem. J. 2004, 383 Pt 1, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Rappu, P.; Lange, S.C.; Mäntsälä, P.; Zalkin, H.; Smith, J.L. Crystal structure of Bacillus subtilis YabJ, a purine regulatory protein and member of the highly conserved YjgF family. Proc. Natl. Acad. Sci. USA 1999, 96, 13074–13079. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Yoshikawa, H.; Shirahige, K. A member of the YER057c/yjgf/Uk114 family links isoleucine biosynthesis and intact mitochondria maintenance in Saccharomyces cerevisiae. Genes Cells 2001, 6, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Leitner-Dagan, Y.; Ovadis, M.; Zuker, A.; Shklarman, E.; Ohad, I.; Tzfira, T.; Vainstein, A. CHRD, a plant member of the evolutionarily conserved YjgF family, influences photosynthesis and chromoplastogenesis. Planta 2006, 225, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Su, P.; Feng, T.; Zhou, X.; Zhang, S.; Zhang, Y.; Cheng, J.; Luo, Y.; Peng, J.; Zhang, Z.; Lu, X.; et al. Isolation of Rhp-PSP, a member of YER057c/YjgF/UK114 protein family with antiviral properties, from the photosynthetic bacterium Rhodopseudomonas palustris strain JSC-3b. Sci. Rep. 2015, 5, 16121. [Google Scholar] [CrossRef] [PubMed]
- Kanouchi, H.; Tachibana, H.; Oka, T.; Yamada, K. Recombinant expression of perchloric acid-soluble protein reduces cell proliferation. Cell. Mol. Life Sci. CMLS 2001, 58, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, T.; Oka, T.; Tokumura, A.; Nishimoto, Y.; Munoz, S.; Kuwahata, M.; Okita, M.; Tsuji, H.; Natori, Y. Analysis of the fatty acid components in a perchloric acid-soluble protein. Biochim. Biophys. Acta 1999, 1437, 317–324. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K.; Mirnikjoo, B.; Schroit, A.J. Regulated externalization of phosphatidylserine at the cell surface: Implications for apoptosis. J. Biol. Chem. 2007, 282, 18357–18364. [Google Scholar] [CrossRef] [PubMed]
- Wyllie, A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Xu, M. DNA fragmentation in apoptosis. Cell Res. 2000, 10, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Tandon, S. DNA fragmentation and cell cycle arrest: A hallmark of apoptosis induced by Ruta graveolens in human colon cancer cells. Homeopathy 2015, 104, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Meng, X.W.; Flatten, K.S.; Loegering, D.A.; Kaufmann, S.H. Phosphatidylserine exposure during apoptosis reflects bidirectional trafficking between plasma membrane and cytoplasm. Cell Death Differ. 2013, 20, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Susin, S.A.; Daugas, E.; Ravagnan, L.; Samejima, K.; Zamzami, N.; Loeffler, M.; Costantini, P.; Ferri, K.F.; Irinopoulou, T.; Prévost, M.C.; et al. Two distinct pathways leading to nuclear apoptosis. J. Exp. Med. 2000, 192, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.M.; Pawlowski, K.; Haas, E.; Ware, C.F.; Godzik, A.; Reed, J.C. A diverse family of proteins containing tumor necrosis factor receptor-associated factor domains. J. Biol. Chem. 2001, 276, 24242–24252. [Google Scholar] [CrossRef] [PubMed]
- Hockenbery, D.; Nunez, G.; Milliman, C.; Schreiber, R.D.; Korsmeyer, S.J. Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 1990, 348, 334–336. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Shi, Y. Molecular mechanisms of caspase regulation during apoptosis. Nature reviews Mol. Cell Biol. 2004, 5, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; McDonnell, J.M.; Korsmeyer, S.J. BCL-2 family members and the mitochondria in apoptosis. Genes Dev. 1999, 13, 1899–1911. [Google Scholar] [CrossRef] [PubMed]
- Naseri, M.H.; Mahdavi, M.; Davoodi, J.; Tackallou, S.H.; Goudarzvand, M.; Neishabouri, S.H. Up regulation of Bax and down regulation of Bcl2 during 3-NC mediated apoptosis in human cancer cells. Cancer Cell Int. 2015, 15, 55. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. The pathophysiology of mitochondrial cell death. Science 2004, 305, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Smaili, S.S.; Hsu, Y.T.; Sanders, K.M.; Russell, J.T.; Youle, R.J. Bax translocation to mitochondria subsequent to a rapid loss of mitochondrial membrane potential. Cell Death Differ. 2001, 8, 909–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-κB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef] [PubMed]
- Frankenberger, M.; Pforte, A.; Sternsdorf, T.; Passlick, B.; Baeuerle, P.A.; Ziegler-Heitbrock, H.W. Constitutive nuclear NF-κB in cells of the monocyte lineage. Biochem. J. 1994, 304 Pt 1, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Han, S.S.; Kim, K.; Hahm, E.R.; Lee, S.J.; Surh, Y.J.; Park, H.K.; Kim, W.S.; Jung, C.W.; Lee, M.H.; Park, K.; et al. l-ascorbic acid represses constitutive activation of NF-κB and COX-2 expression in human acute myeloid leukemia, HL-60. J. Cell. Biochem. 2004, 93, 257–270. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-κB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, Z.; Akbarzadeh, A.; Rahmatiyamchi, M.; Movassaghpour, A.A.; Alipour, M.; Nejati-Koshki, K.; Sadeghi, Z.; Dariushnejad, H.; Zarghami, N. Molecular target therapy of AKT and NF-κB signaling pathways and multidrug resistance by specific cell penetrating inhibitor peptides in HL-60 cells. Asian Pac. J. Cancer Prev. APJCP 2014, 15, 4353–4358. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-L.; Kamata, H.; Karin, M. IKK/NF-κB signaling: Balancing life and death—A new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.N.; Koo, K.H.; Sung, J.Y.; Yun, U.J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhong, L.; Liu, B.Z.; Gao, Y.J.; Gao, Y.M.; Hu, X.X. Effect of GINS2 on proliferation and apoptosis in leukemic cell line. Int. J. Med. Sci. 2013, 10, 1795–1804. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Grana, X.; Reddy, E.P. Cell cycle control in mammalian cells: Role of cyclins, cyclin dependent kinases (CDKs), growth suppressor genes and cyclin-dependent kinase inhibitors (CKIs). Oncogene 1995, 11, 211–219. [Google Scholar] [PubMed]
- Kastan, M.B.; Canman, C.E.; Leonard, C.J. P53, cell cycle control and apoptosis: Implications for cancer. Cancer Metastasis Rev. 1995, 14, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ching, Y.Q.; Chng, W.J. Aberrant nuclear factor-κB activity in acute myeloid leukemia: From molecular pathogenesis to therapeutic target. Oncotarget 2015, 6, 5490–5500. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.E.; Huang, D.B.; Chen, Y.Q.; Ghosh, G. Crystal structure of p50/p65 heterodimer of transcription factor NF-κB bound to DNA. Nature 1998, 391, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-κB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Liptay, S.; Weber, C.K.; Ludwig, L.; Wagner, M.; Adler, G.; Schmid, R.M. Mitogenic and antiapoptotic role of constitutive NF-κB/Rel activity in pancreatic cancer. Int. J. Cancer 2003, 105, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Nuclear factor-κB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Sliva, D. Signaling Pathways Responsible for Cancer Cell Invasion as Targets for Cancer Therapy. Curr. Cancer Drug Targets 2004, 4, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Joseph, T.P.; Chanda, W.; Padhiar, A.A.; Batool, S.; LiQun, S.; Zhong, M.; Huang, M. A Preclinical Evaluation of the Antitumor Activities of Edible and Medicinal Mushrooms: A Molecular Insight. Integr. Cancer Ther. 2017. [Google Scholar] [CrossRef] [PubMed]
- Colombo, I.; Ceciliani, F.; Ronchi, S.; Bartorelli, A.; Berra, B. cDNA cloning and Escherichia coli expression of UK114 tumor antigen. Biochim. Biophys. Acta 1998, 1442, 49–59. [Google Scholar] [CrossRef]
- Ardelt, B.; Ardelt, W.; Pozarowski, P.; Kunicki, J.; Shogen, K.; Darzynkiewicz, Z. Cytostatic and cytotoxic properties of Amphinase: A novel cytotoxic ribonuclease from Rana pipiens oocytes. Cell Cycle Georget. Tex 2007, 6, 3097–3102. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, W.; Shogen, K.; Darzynkiewicz, Z. Onconase and amphinase, the antitumor ribonucleases from Rana pipiens oocytes. Curr. Pharm. Biotechnol. 2008, 9, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Ribo, M.; Navarro, S.; Nogues, M.V.; Vilanova, M.; Benito, A. A human ribonuclease induces apoptosis associated with p21WAF1/CIP1 induction and JNK inactivation. BMC Cancer 2011, 11, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhao, L.; Wang, H.; Ng, T.B. A novel ribonuclease with antiproliferative activity toward leukemia and lymphoma cells and HIV-1 reverse transcriptase inhibitory activity from the mushroom, Hohenbuehelia serotina. Int. J. Mol. Med. 2014, 33, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Vert, A.; Castro, J.; Ribo, M.; Benito, A.; Vilanova, M. A nuclear-directed human pancreatic ribonuclease (PE5) targets the metabolic phenotype of cancer cells. Oncotarget 2016, 7, 18309–18324. [Google Scholar] [CrossRef] [PubMed]
- McKenna, S.A.; Lindhout, D.A.; Kim, I.; Liu, C.W.; Gelev, V.M.; Wagner, G.; Puglisi, J.D. Molecular framework for the activation of RNA-dependent protein kinase. J. Biol. Chem. 2007, 282, 11474–11486. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Gotte, G.; Picone, D.; Donadelli, M. Onconase induces autophagy sensitizing pancreatic cancer cells to gemcitabine and activates Akt/mTOR pathway in a ROS-dependent manner. Biochim. Biophys. Acta BBA Mol. Cell Res. 2015, 1853, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, B.; Juan, G.; Burfeind, P.; Salomon, T.; Wu, J.M.; Hsieh, T.C.; Li, X.; Sperry, R.; Pozarowski, P.; Shogen, K.; et al. Onconase, an anti-tumor ribonuclease suppresses intracellular oxidative stress. Int. J. Oncol. 2007, 31, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Yang, J.S.; Huang, A.C.; Hsia, T.C.; Chou, S.T.; Kuo, C.L.; Lu, H.F.; Lee, T.H.; Wood, W.G.; Chung, J.G. Chrysophanol induces necrosis through the production of ROS and alteration of ATP levels in J5 human liver cancer cells. Mol. Nutr. Food Res. 2010, 54, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Im, M.; Yim, N.H.; Ma, J.Y. Reduction of metastatic and angiogenic potency of malignant cancer by Eupatorium fortunei via suppression of MMP-9 activity and VEGF production. Sci. Rep. 2014, 4, 6994. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Forward 5′ → 3′ | Reverse 5′ → 3′ |
|---|---|---|
| Bax | CCCGAGAGGTCTTTTTCCGAG | CCAGCCCATGATGGTTCTGAT |
| Bcl-2 | GGTGGGGTCATGTGTGTGG | CGGTTCAGGTACTCAGTCATCC |
| Cas3 | AGAGGGGATCGTTGTAGAAGTC | ACAGTCCAGTTCTGTACCACG |
| Cas8 | TTTCTGCCTACAGGGTCATGC | TGTCCAACTTTCCTTCTCCCA |
| Cas9 | CTCAGACCAGAGATTCGCAAAC | GCATTTCCCCTCAAACTCTCAA |
| Cdk2 | GCCATTCTCATCGGGTCCTC | ATTTGCAGCCCAGGAGGATT |
| Cdk4 | ATGGCTACCTCTCGATATGAGC | CATTGGGGACTCTCACACTCT |
| Cdk6 | CTGCAGGGAAAGAAAAGTGC | CTCCTCGAAGCGAAGTCCTC |
| Cyclin D1 | GCTGCGAAGTGGAAACCAT | CCTCCTTCTGCACACATTTGAA |
| Cyclin E1 | AAGGAGCGGGACACCATGA | ACGGTCACGTTTGCCTTCC |
| FasL | TGCCTTGGTAGGATTGGGC | GCTGGTAGACTCTCGGAGTTC |
| GAPDH | AATCCCATCACCATCTTCCA | TGGACTCCACGACGTACTCA |
| p21 | TGTCCGTCAGAACCCATGC | AAAGTCGAAGTTCCATCGCTC |
| p27 | TAATTGGGGCTCCGGCTAACT | TGCAGGTCGCTTCCTTATTCC |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joseph, T.P.; Chanda, W.; Mohammad, A.F.; Kanwal, S.; Batool, S.; Zhang, M.; Zhong, M.; Huang, M. Lp16-PSP, a Member of YjgF/YER057c/UK114 Protein Family Induces Apoptosis and p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in Human Acute Promyelocytic Leukemia (APL) HL-60 Cells. Int. J. Mol. Sci. 2017, 18, 2407. https://doi.org/10.3390/ijms18112407
Joseph TP, Chanda W, Mohammad AF, Kanwal S, Batool S, Zhang M, Zhong M, Huang M. Lp16-PSP, a Member of YjgF/YER057c/UK114 Protein Family Induces Apoptosis and p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in Human Acute Promyelocytic Leukemia (APL) HL-60 Cells. International Journal of Molecular Sciences. 2017; 18(11):2407. https://doi.org/10.3390/ijms18112407
Chicago/Turabian StyleJoseph, Thomson Patrick, Warren Chanda, Abdullah Faqeer Mohammad, Sadia Kanwal, Samana Batool, Meishan Zhang, Mintao Zhong, and Min Huang. 2017. "Lp16-PSP, a Member of YjgF/YER057c/UK114 Protein Family Induces Apoptosis and p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in Human Acute Promyelocytic Leukemia (APL) HL-60 Cells" International Journal of Molecular Sciences 18, no. 11: 2407. https://doi.org/10.3390/ijms18112407
APA StyleJoseph, T. P., Chanda, W., Mohammad, A. F., Kanwal, S., Batool, S., Zhang, M., Zhong, M., & Huang, M. (2017). Lp16-PSP, a Member of YjgF/YER057c/UK114 Protein Family Induces Apoptosis and p21WAF1/CIP1 Mediated G1 Cell Cycle Arrest in Human Acute Promyelocytic Leukemia (APL) HL-60 Cells. International Journal of Molecular Sciences, 18(11), 2407. https://doi.org/10.3390/ijms18112407