Non-Catalytic Roles of the Topoisomerase IIα C-Terminal Domain


Abstract
: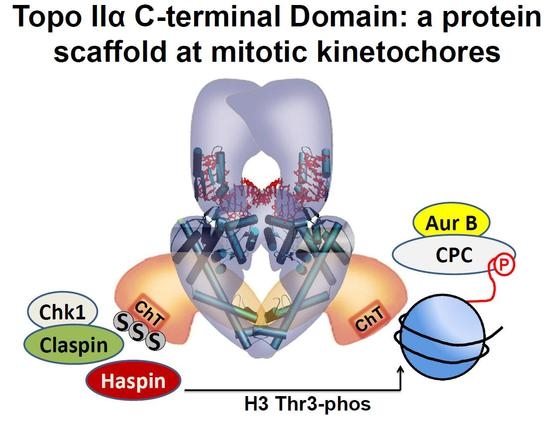
{kind=link}
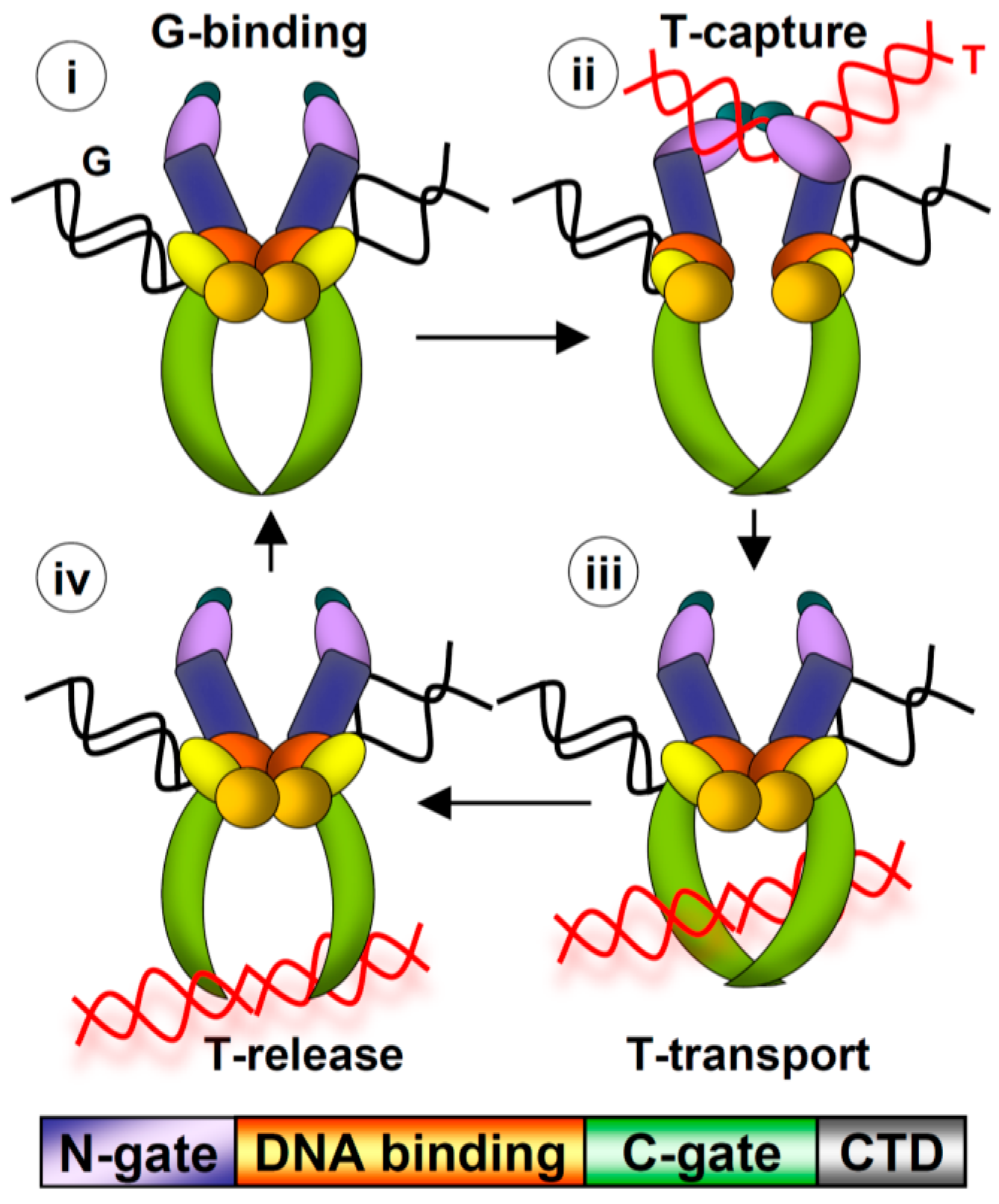{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Evidence That the CTD Mediates Functional Interactions with Chromatin
3. The Topo IIα CTD Serves as a Scaffold to Recruit Mitotic Regulators to Centromeres
4. Functions of the CTD at Yeast Centromeres
5. Evidence That the CTD of Budding Yeast Top2 Functions in Checkpoint Signaling
6. Prospective Questions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Topo IIα | Topoisomerase IIα |
| Topo IIβ | Topoisomerase IIβ |
| CTD | C-terminal domain |
| UFB | Ultra-fine DNA bridge |
| XEE | Xenopus egg extract |
| GFP | Green fluorescent protein |
| ChT | Chromatin Tether |
| FRAP | Fluorescence recovery after photo-bleaching |
| H3T3-Phos | Phosphorylated threonine 3 residue of histone H3 |
| CPC | Chromosome Passenger Complex |
| SAC | Spindle assembly checkpoint |
| NPC | Nuclear Pore Complex |
| TER | Site of DNA replication termination |
| STUbL | SUMO-targeted ubiquitin ligase |
References
- Grue, P.; Grasser, A.; Sehested, M.; Jensen, P.B.; Uhse, A.; Straub, T.; Ness, W.; Boege, F. Essential mitotic functions of DNA topoisomerase IIalpha are not adopted by topoisomerase IIbeta in human H69 cells. J. Biol. Chem. 1998, 273, 33660–33666. [Google Scholar] [CrossRef] [PubMed]
- Linka, R.M.; Porter, A.C.; Volkov, A.; Mielke, C.; Boege, F.; Christensen, M.O. C-terminal regions of topoisomerase IIalpha and IIbeta determine isoform-specific functioning of the enzymes in vivo. Nucleic Acids Res. 2007, 35, 3810–3822. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Miyaike, M.; Kato, S.; Kanamaru, R.; Koyama, H.; Kikuchi, A. Cellular distribution of mammalian DNA topoisomerase II is determined by its catalytically dispensable C-terminal domain. Nucleic Acids Res. 1997, 25, 3135–3142. [Google Scholar] [CrossRef] [PubMed]
- Dickey, J.S.; Osheroff, N. Impact of the C-terminal domain of topoisomerase IIalpha on the DNA cleavage activity of the human enzyme. Biochemistry 2005, 44, 11546–11554. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell Biol. 2003, 163, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Bachant, J.; Alcasabas, A.; Blat, Y.; Kleckner, N.; Elledge, S.J. The SUMO-1 isopeptidase Smt4 is linked to centromeric cohesion through SUMO-1 modification of DNA topoisomerase II. Mol. Cell 2002, 9, 1169–1182. [Google Scholar] [CrossRef]
- Lane, A.B.; Gimenez-Abian, J.F.; Clarke, D.J. A novel chromatin tether domain controls topoisomerase IIalpha dynamics and mitotic chromosome formation. J. Cell Biol. 2013, 203, 471–486. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Halligan, B.; Cooke, C.A.; Heck, M.M.; Liu, L.F. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J. Cell Biol. 1985, 100, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Heck, M.M. Localization of topoisomerase II in mitotic chromosomes. J. Cell Biol. 1985, 100, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Gasser, S.M.; Laroche, T.; Falquet, J.; Boy de la Tour, E.; Laemmli, U.K. Metaphase chromosome structure. Involvement of topoisomerase II. J. Mol. Biol. 1986, 188, 613–629. [Google Scholar] [CrossRef]
- Warburton, P.E.; Earnshaw, W.C. Untangling the role of DNA topoisomerase II in mitotic chromosome structure and function. Bioessays 1997, 19, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Mitchison, T.J. A heterodimeric coiled-coil protein required for mitotic chromosome condensation in vitro. Cell 1994, 79, 449–458. [Google Scholar] [CrossRef]
- Hirano, T.; Mitchison, T.J. Topoisomerase II does not play a scaffolding role in the organization of mitotic chromosomes assembled in Xenopus egg extracts. J. Cell Biol. 1993, 120, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Abian, J.F.; Clarke, D.J.; Mullinger, A.M.; Downes, C.S.; Johnson, R.T. A postprophase topoisomerase II-dependent chromatid core separation step in the formation of metaphase chromosomes. J. Cell Biol. 1995, 131, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.A.; Come, M.G.; Hudson, J.R.; Mo, Y.Y.; Beck, W.T.; Gorbsky, G.J. Rapid exchange of mammalian topoisomerase II alpha at kinetochores and chromosome arms in mitosis. J. Cell Biol. 2002, 158, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Laemmli, U.K. A two-step scaffolding model for mitotic chromosome assembly. Dev. Cell 2003, 4, 467–480. [Google Scholar] [CrossRef]
- Christensen, M.O.; Larsen, M.K.; Barthelmes, H.U.; Hock, R.; Andersen, C.L.; Kjeldsen, E.; Knudsen, B.R.; Westergaard, O.; Boege, F.; Mielke, C. Dynamics of human DNA topoisomerases IIalpha and IIbeta in living cells. J. Cell Biol. 2002, 157, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Swedlow, J.R.; Sedat, J.W.; Agard, D.A. Multiple chromosomal populations of topoisomerase II detected in vivo by time-lapse, three-dimensional wide-field microscopy. Cell 1993, 73, 97–108. [Google Scholar] [CrossRef]
- Gilroy, K.L.; Austin, C.A. The impact of the C-terminal domain on the interaction of human DNA topoisomerase II alpha and beta with DNA. PLoS ONE 2011, 6, e14693. [Google Scholar] [CrossRef] [PubMed]
- Meczes, E.L.; Gilroy, K.L.; West, K.L.; Austin, C.A. The impact of the human DNA topoisomerase II C-terminal domain on activity. PLoS ONE 2008, 3, e1754. [Google Scholar] [CrossRef] [PubMed]
- Luo, K.; Yuan, J.; Chen, J.; Lou, Z. Topoisomerase IIalpha controls the decatenation checkpoint. Nat. Cell Biol. 2009, 11, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.N.; Kjeldsen, E.; Straub, T.; Knudsen, B.R.; Hickson, I.D.; Kikuchi, A.; Kreipe, H.; Boege, F. Cell cycle-coupled relocation of types I and II topoisomerases and modulation of catalytic enzyme activities. J. Cell Biol. 1997, 136, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Furuta, M.; Kirkpatrick, D.; Gygi, S.P.; Azuma, Y. PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J. Cell Biol. 2010, 191, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Martinez, L.A.; Gimenez-Abian, J.F.; Azuma, Y.; Guacci, V.; Gimenez-Martin, G.; Lanier, L.M.; Clarke, D.J. PIASgamma is required for faithful chromosome segregation in human cells. PLoS ONE 2006, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B.; Kao, E.; Sustmann, C.; Tahk, S.; Shuai, K.; Grosschedl, R.; van Deursen, J.M. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, S.; Psakhye, I. Control of nuclear activities by substrate-selective and protein-group SUMOylation. Annu. Rev. Genet. 2013, 47, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Farr, C.J.; Antoniou-Kourounioti, M.; Mimmack, M.L.; Volkov, A.; Porter, A.C. The alpha isoform of topoisomerase II is required for hypercompaction of mitotic chromosomes in human cells. Nucleic Acids Res. 2014, 42, 4414–4426. [Google Scholar] [CrossRef] [PubMed]
- Gimenez-Abian, J.F.; Clarke, D.J. Cytological analysis of chromosome structural defects that result from topoisomerase II dysfunction. Methods Mol. Biol. 2009, 582, 189–207. [Google Scholar] [PubMed]
- Wei, Y.; Mizzen, C.A.; Cook, R.G.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc. Natl. Acad. Sci. USA 1998, 95, 7480–7484. [Google Scholar] [CrossRef] [PubMed]
- Van Hooser, A.; Goodrich, D.W.; Allis, C.D.; Brinkley, B.R.; Mancini, M.A. Histone H3 phosphorylation is required for the initiation, but not maintenance, of mammalian chromosome condensation. J. Cell Sci. 1998, 111, 3497–3506. [Google Scholar] [PubMed]
- Wei, Y.; Yu, L.; Bowen, J.; Gorovsky, M.A.; Allis, C.D. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell 1999, 97, 99–109. [Google Scholar] [CrossRef]
- Edgerton, H.; Johansson, M.; Keifenheim, D.; Mukherjee, S.; Chacon, J.M.; Bachant, J.; Gardner, M.K.; Clarke, D.J. A noncatalytic function of the topoisomerase II CTD in Aurora B recruitment to inner centromeres during mitosis. J. Cell Biol. 2016, 213, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.M.; Ting, L.; Gygi, S.P.; Azuma, Y. SUMOylation of DNA topoisomerase IIalpha regulates histone H3 kinase Haspin and H3 phosphorylation in mitosis. J. Cell Biol. 2016, 213, 665–678. [Google Scholar] [CrossRef] [PubMed]
- De la Barre, A.E.; Gerson, V.; Gout, S.; Creaven, M.; Allis, C.D.; Dimitrov, S. Core histone N-termini play an essential role in mitotic chromosome condensation. EMBO J. 2000, 19, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.M.; Fournier, R.E.; Oshimura, M.; Regnier, V.; Farr, C.J. Topoisomerase II cleavage activity within the human D11Z1 and DXZ1 alpha-satellite arrays. Chromosome Res. 2005, 13, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Wandall, A.; Kjeldsen, E.; Mielke, C.; Koch, J. Active, but not inactive, human centromeres display topoisomerase II activity in vivo. Chromosome Res. 2002, 10, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.M.; Critcher, R.; Ebersole, T.A.; Valdivia, M.M.; Earnshaw, W.C.; Fukagawa, T.; Farr, C.J. Co-localization of centromere activity, proteins and topoisomerase II within a subdomain of the major human X alpha-satellite array. EMBO J. 2002, 21, 5269–5280. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Yoshida, M.M.; Sridharan, V.; Kumagai, A.; Dunphy, W.G.; Dasso, M.; Azuma, Y. SUMOylation of the C-terminal domain of DNA topoisomerase IIα regulates the centromeric localization of Claspin. Cell Cycle 2015, 14, 2777–2784. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Arnaoutov, A.; Anan, T.; Dasso, M. PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J. 2005, 24, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Al-Ani, G.; Deckert, K.; Kirkpatrick, D.; Gygi, S.P.; Dasso, M.; Azuma, Y. PIASy mediates SUMO-2/3 conjugation of poly(ADP-ribose) polymerase 1 (PARP1) on mitotic chromosomes. J. Biol. Chem. 2010, 285, 14415–14423. [Google Scholar] [CrossRef] [PubMed]
- Petsalaki, E.; Akoumianaki, T.; Black, E.J.; Gillespie, D.A.; Zachos, G. Phosphorylation at serine 331 is required for Aurora B activation. J. Cell Biol. 2011, 195, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.E.; Ghenoiu, C.; Xue, J.Z.; Zierhut, C.; Kimura, H.; Funabiki, H. Survivin reads phosphorylated histone H3 threonine 3 to activate the mitotic kinase Aurora B. Science 2010, 330, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Jeyaprakash, A.A.; Basquin, C.; Jayachandran, U.; Conti, E. Structural basis for the recognition of phosphorylated histone h3 by the survivin subunit of the chromosomal passenger complex. Structure 2011, 19, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Carmena, M.; Wheelock, M.; Funabiki, H.; Earnshaw, W.C. The chromosomal passenger complex (CPC): From easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol. 2012, 13, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Redwood, C.S.; Jenkins, J.R.; Andersen, A.H.; Hickson, I.D. Human DNA topoisomerases II alpha and II beta can functionally substitute for yeast TOP2 in chromosome segregation and recombination. Mol. Gen. Genet. 1996, 252, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Rattner, J.B.; Hendzel, M.J.; Furbee, C.S.; Muller, M.T.; Bazett-Jones, D.P. Topoisomerase II alpha is associated with the mammalian centromere in a cell cycle- and species-specific manner and is required for proper centromere/kinetochore structure. J. Cell Biol. 1996, 134, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J.; Goulding, S.; Earnshaw, W.C.; Carmena, M. RNAi analysis reveals an unexpected role for topoisomerase II in chromosome arm congression to a metaphase plate. J. Cell Sci. 2003, 116, 4715–4726. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.C.; Farr, C.J. Topoisomerase II: Untangling its contribution at the centromere. Chromosome Res. 2004, 12, 569–583. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, Y.; Yanagida, M. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol. Biol. Cell 2006, 17, 2287–2302. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.J.; Vas, A.C.; Andrews, C.A.; Diaz-Martinez, L.A.; Gimenez-Abian, J.F. Topoisomerase II checkpoints: Universal mechanisms that regulate mitosis. Cell Cycle 2006, 5, 1925–1928. [Google Scholar] [CrossRef] [PubMed]
- Skoufias, D.A.; Lacroix, F.B.; Andreassen, P.R.; Wilson, L.; Margolis, R.L. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol. Cell 2004, 15, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.A.; Vas, A.C.; Meier, B.; Gimenez-Abian, J.F.; Diaz-Martinez, L.A.; Green, J.; Erickson, S.L.; Vanderwaal, K.E.; Hsu, W.S.; Clarke, D.J. A mitotic topoisomerase II checkpoint in budding yeast is required for genome stability but acts independently of Pds1/securin. Genes Dev. 2006, 20, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Furniss, K.L.; Tsai, H.J.; Byl, J.A.; Lane, A.B.; Vas, A.C.; Hsu, W.S.; Osheroff, N.; Clarke, D.J. Direct monitoring of the strand passage reaction of DNA topoisomerase II triggers checkpoint activation. PLoS Genet. 2013, 9, e1003832. [Google Scholar] [CrossRef] [PubMed]
- Chatel, G.; Fahrenkrog, B. Nucleoporins: Leaving the nuclear pore complex for a successful mitosis. Cell Signal. 2011, 23, 1555–1562. [Google Scholar] [CrossRef] [PubMed]
- Iouk, T.; Kerscher, O.; Scott, R.J.; Basrai, M.A.; Wozniak, R.W. The yeast nuclear pore complex functionally interacts with components of the spindle assembly checkpoint. J. Cell Biol. 2002, 159, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.J.; Lusk, C.P.; Dilworth, D.J.; Aitchison, J.D.; Wozniak, R.W. Interactions between Mad1p and the nuclear transport machinery in the yeast Saccharomyces cerevisiae. Mol. Biol. Cell 2005, 16, 4362–4374. [Google Scholar] [CrossRef] [PubMed]
- Goh, P.Y.; Kilmartin, J.V. NDC10: A gene involved in chromosome segregation in Saccharomyces cerevisiae. J. Cell Biol. 1993, 121, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.A.; Burke, D.J. Cell cycle arrest in cdc20 mutants of Saccharomyces cerevisiae is independent of Ndc10p and kinetochore function but requires a subset of spindle checkpoint genes. Genetics 1998, 148, 1701–1713. [Google Scholar] [PubMed]
- Rodriguez-Bravo, V.; Maciejowski, J.; Corona, J.; Buch, H.K.; Collin, P.; Kanemaki, M.T.; Shah, J.V.; Jallepalli, P.V. Nuclear pores protect genome integrity by assembling a premitotic and Mad1-dependent anaphase inhibitor. Cell 2014, 156, 1017–1031. [Google Scholar] [CrossRef] [PubMed]
- Baxter, J.; Diffley, J.F. Topoisomerase II inactivation prevents the completion of DNA replication in budding yeast. Mol. Cell 2008, 30, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Phua, H.H.; Bennett, S.C.; Spence, J.M.; Farr, C.J. Studying vertebrate topoisomerase 2 function using a conditional knockdown system in DT40 cells. Nucleic Acids Res. 2009, 37, e98. [Google Scholar] [CrossRef] [PubMed]
- Bower, J.J.; Karaca, G.F.; Zhou, Y.; Simpson, D.A.; Cordeiro-Stone, M.; Kaufmann, W.K. Topoisomerase IIalpha maintains genomic stability through decatenation G(2) checkpoint signaling. Oncogene 2010, 29, 4787–4799. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Fachinetti, D.; Bermejo, R.; Cocito, A.; Minardi, S.; Katou, Y.; Kanoh, Y.; Shirahige, K.; Azvolinsky, A.; Zakian, V.A.; Foiani, M. Replication termination at eukaryotic chromosomes is mediated by Top2 and occurs at genomic loci containing pausing elements. Mol. Cell 2010, 39, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Comings, D.E. Arrangement of chromatin in the nucleus. Hum. Genet. 1980, 53, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Jaunin, F.; Fakan, S. DNA replication and nuclear architecture. J. Cell Biochem. 2002, 85, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Agostinho, M.; Rino, J.; Braga, J.; Ferreira, F.; Steffensen, S.; Ferreira, J. Human topoisomerase IIalpha: Targeting to subchromosomal sites of activity during interphase and mitosis. Mol. Biol. Cell 2004, 15, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Winey, M.; Yarar, D.; Giddings, T.H., Jr.; Mastronarde, D.N. Nuclear pore complex number and distribution throughout the Saccharomyces cerevisiae cell cycle by three-dimensional reconstruction from electron micrographs of nuclear envelopes. Mol. Biol. Cell 1997, 8, 2119–2132. [Google Scholar] [CrossRef] [PubMed]
- Niepel, M.; Molloy, K.R.; Williams, R.; Farr, J.C.; Meinema, A.C.; Vecchietti, N.; Cristea, I.M.; Chait, B.T.; Rout, M.P.; Strambio-De-Castillia, C. The nuclear basket proteins Mlp1p and Mlp2p are part of a dynamic interactome including Esc1p and the proteasome. Mol. Biol. Cell 2013, 24, 3920–3938. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Schwarzbraun, T.; Speicher, M.R.; Nigg, E.A. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma 2008, 117, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.F.; Huttner, D.; Bizard, A.H.; Hirano, S.; Li, T.N.; Palmai-Pallag, T.; Bjerregaard, V.A.; Liu, Y.; Nigg, E.A.; Wang, L.H.; et al. PICH promotes sister chromatid disjunction and co-operates with topoisomerase II in mitosis. Nat. Commun. 2015, 6, 8962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sridharan, V.; Park, H.; Ryu, H.; Azuma, Y. SUMOylation regulates polo-like kinase 1-interacting checkpoint helicase (PICH) during mitosis. J. Biol. Chem. 2015, 290, 3269–3276. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, V.; Azuma, Y. SUMO-interacting motifs (SIMs) in Polo-like kinase 1-interacting checkpoint helicase (PICH) ensure proper chromosome segregation during mitosis. Cell Cycle 2016, 15, 2135–2144. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Desai, S.D.; Ting, C.Y.; Hwang, J.; Liu, L.F. 26 S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 2001, 276, 40652–40658. [Google Scholar] [CrossRef] [PubMed]
- Isik, S.; Sano, K.; Tsutsui, K.; Seki, M.; Enomoto, T.; Saitoh, H.; Tsutsui, K. The SUMO pathway is required for selective degradation of DNA topoisomerase IIbeta induced by a catalytic inhibitor ICRF-193(1). FEBS Lett. 2003, 546, 374–378. [Google Scholar] [CrossRef]
- Agostinho, M.; Santos, V.; Ferreira, F.; Costa, R.; Cardoso, J.; Pinheiro, I.; Rino, J.; Jaffray, E.; Hay, R.T.; Ferreira, J. Conjugation of human topoisomerase 2α with small ubiquitin-like modifiers 2/3 in response to topoisomerase inhibitors: Cell cycle stage and chromosome domain specificity. Cancer Res. 2008, 68, 2409–2418. [Google Scholar] [CrossRef] [PubMed]
- Onoda, A.; Hosoya, O.; Sano, K.; Kiyama, K.; Kimura, H.; Kawano, S.; Furuta, R.; Miyaji, M.; Tsutsui, K.; Tsutsui, K.M. Nuclear dynamics of topoisomerase IIbeta reflects its catalytic activity that is regulated by binding of RNA to the C-terminal domain. Nucleic Acids Res. 2014, 42, 9005–9020. [Google Scholar] [CrossRef] [PubMed]
- Rzepecki, R.; Fisher, P.A. During both interphase and mitosis, DNA topoisomerase II interacts with DNA as well as RNA through the protein’s C-terminal domain. J. Cell Sci. 2000, 113, 1635–1647. [Google Scholar] [PubMed]
- Wotton, D.; Pemberton, L.F.; Merrill-Schools, J. SUMO and Chromatin Remodeling. Adv. Exp. Med. Biol. 2017, 963, 35–50. [Google Scholar] [PubMed]
- Uuskula-Reimand, L.; Hou, H.; Samavarchi-Tehrani, P.; Rudan, M.V.; Liang, M.; Medina-Rivera, A.; Mohammed, H.; Schmidt, D.; Schwalie, P.; Young, E.J.; et al. Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 2016, 17, 182. [Google Scholar] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.K.; Burger, L.; Nikoletopoulou, V.; Deogracias, R.; Thakurela, S.; Wirbelauer, C.; Kaut, J.; Terranova, R.; Hoerner, L.; Mielke, C.; et al. Target genes of Topoisomerase IIbeta regulate neuronal survival and are defined by their chromatin state. Proc. Natl. Acad. Sci. USA 2012, 109, E934–E943. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clarke, D.J.; Azuma, Y. Non-Catalytic Roles of the Topoisomerase IIα C-Terminal Domain. Int. J. Mol. Sci. 2017, 18, 2438. https://doi.org/10.3390/ijms18112438
Clarke DJ, Azuma Y. Non-Catalytic Roles of the Topoisomerase IIα C-Terminal Domain. International Journal of Molecular Sciences. 2017; 18(11):2438. https://doi.org/10.3390/ijms18112438
Chicago/Turabian StyleClarke, Duncan J., and Yoshiaki Azuma. 2017. "Non-Catalytic Roles of the Topoisomerase IIα C-Terminal Domain" International Journal of Molecular Sciences 18, no. 11: 2438. https://doi.org/10.3390/ijms18112438
APA StyleClarke, D. J., & Azuma, Y. (2017). Non-Catalytic Roles of the Topoisomerase IIα C-Terminal Domain. International Journal of Molecular Sciences, 18(11), 2438. https://doi.org/10.3390/ijms18112438