The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC)


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Pathogenesis of Chronic Liver Disease and Cirrhosis
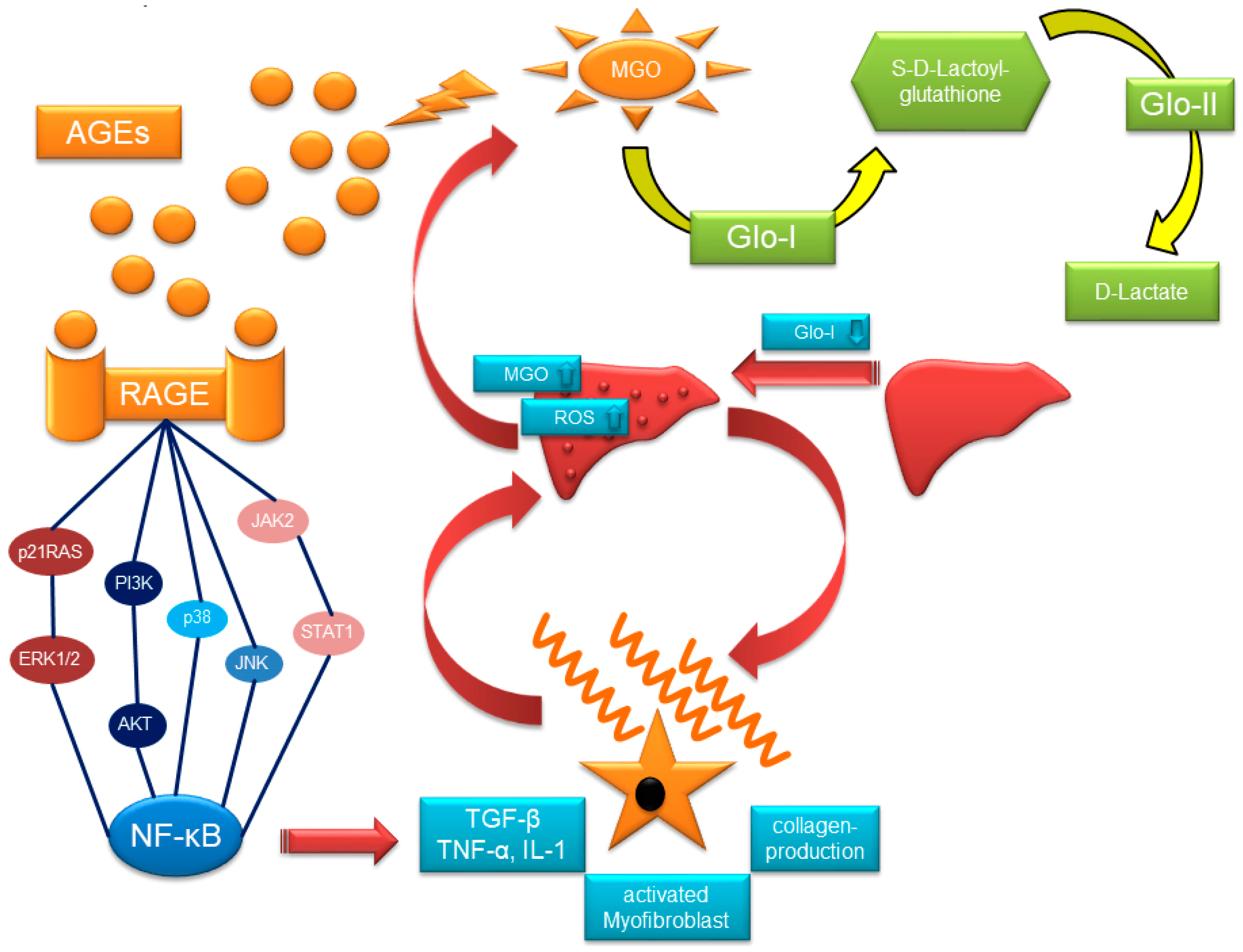
2. Glyoxalase-System, MGO, AGEs and RAGE
2.1. The Glyoxalase-System
2.2. MGO and GO
2.3. RAGE and AGE-Rs
2.4. Clinical Implication of Glo-I
3. Glo-I, AGEs, and RAGE in Chronic Liver Injury
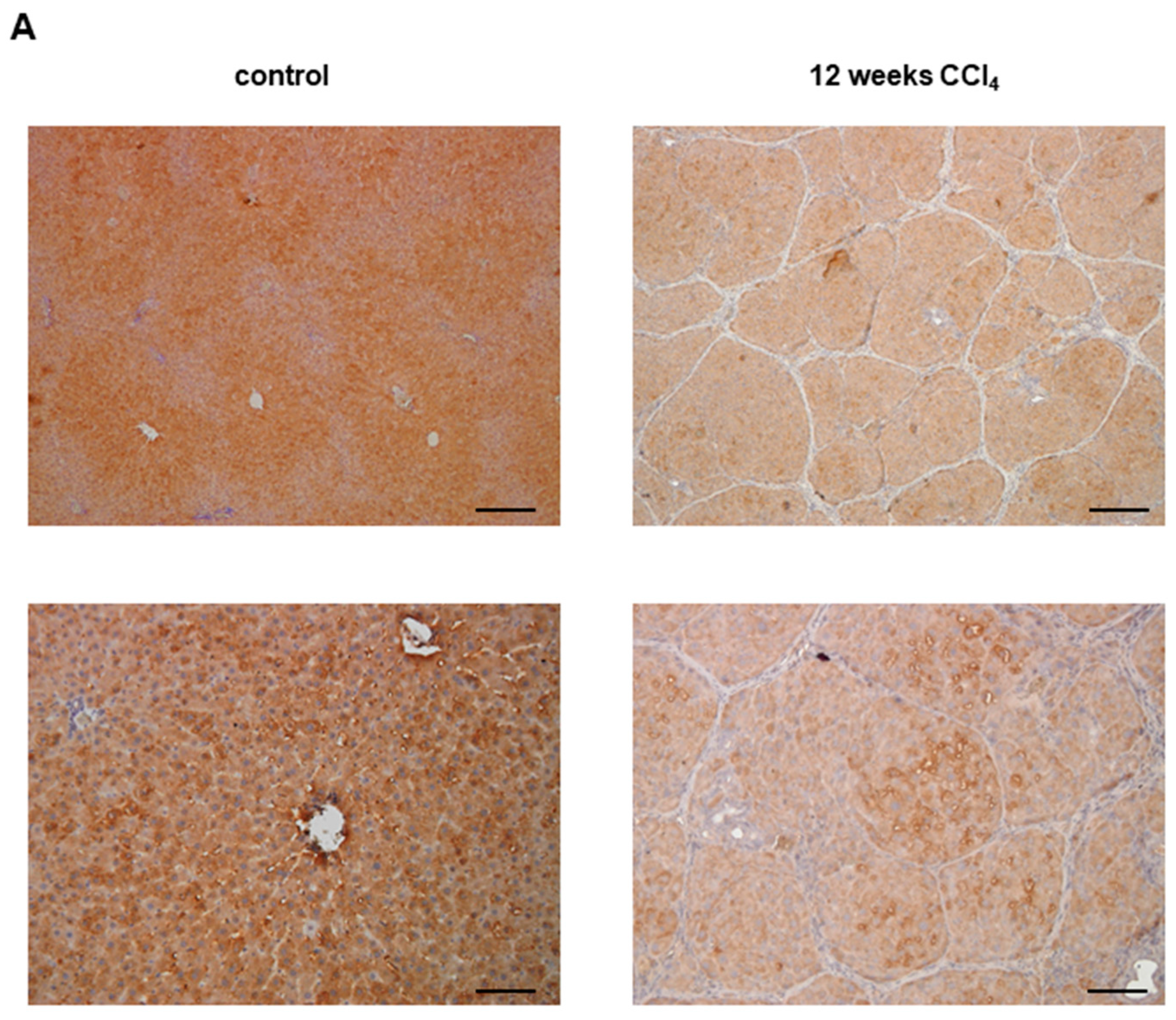
3.1. Glo-I in Liver Disease
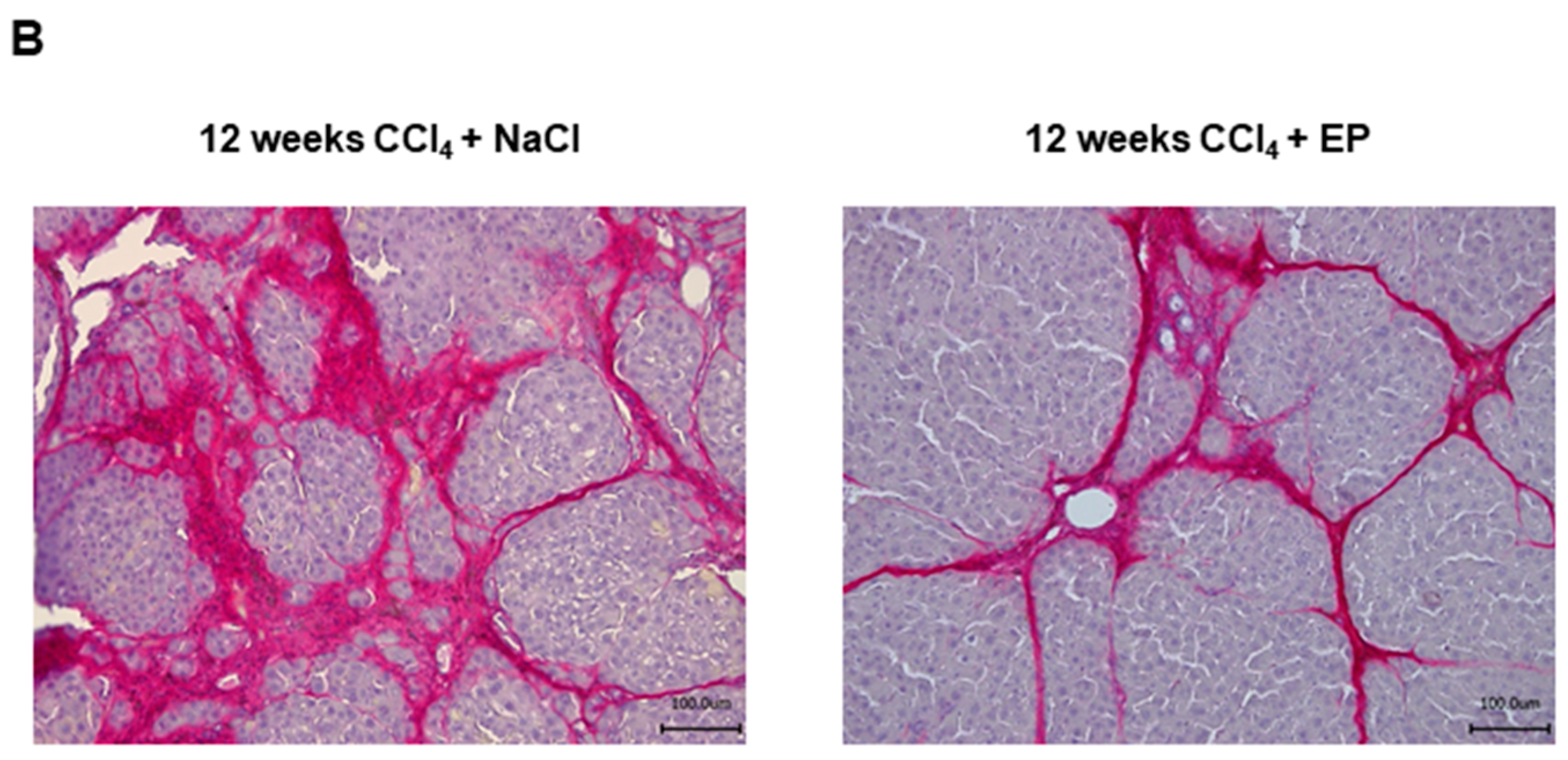
3.2. Effects of Glo-I Modulation by Ethyl Pyruvate
3.3. AGEs in Liver Disease
3.4. RAGE in Liver Disease
4. Glo-I/AGE/RAGE in HCC
Acknowledgments
Conflicts of Interest
Abbreviations
| AGEs | Advanced glycation endproducts |
| EP | Ethyl pyruvate |
| Glo-I | Glyoxalase-I |
| Glo-II | Glyoxalase-II |
| G-H1 | Nδ-(5-hydro-4-imidazolon-2-yl)-ornithine |
| GO | Glyoxal |
| GOLD | Glyoxal lysine dimer |
| GSH | l-glutathione |
| HCC | Hepatocellular carcinoma |
| HEP | Hepatocytes |
| HSC | Hepatic stellate cells |
| KC | Kupffer cells |
| LSEC | Liver sinusoidal endothelial cells |
| MG-H1 | Nδ-(5-hydro-5-methyl-4-imidazolon-2-yl)-ornithine |
| MGO | Methylglyoxal |
| MOLD | Methylglyoxal lysine dimer |
| miRNA | microRNA |
| NAFLD/NASH | Non-alcoholic fatty liver disease/steatohepatitis |
| RAGE | Receptor for advanced glycation endproducts |
| sRAGE | Soluble form of RAGE |
| ROS | Reactive oxygen species |
References
- Rehm, J.; Samokhvalov, A.V.; Shield, K.D. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Cannito, S.; Paternostro, C.; Bocca, C.; Miglietta, A.; Parola, M. Cellular and molecular mechanisms in liver fibrogenesis. Arch. Biochem. Biophys. 2014, 548, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J. Vascular deterioration in cirrhosis: The big picture. J. Clin. Gastroenterol. 2007, 41 (Suppl. S3), S247–S253. [Google Scholar] [CrossRef] [PubMed]
- Groszmann, R.J. Nitric oxide and hemodynamic impairment. Digestion 1998, 59 (Suppl. S2), 6–7. [Google Scholar] [CrossRef] [PubMed]
- Groszmann, R.J. Hyperdynamic circulation of liver disease 40 years later: Pathophysiology and clinical consequences. Hepatology 1994, 20, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Williams, E.; Cadenas, E. Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 2001, 353, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide Ion: Generation and Chemical Implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed]
- Breitkopf, K.; Nagy, L.E.; Beier, J.I.; Mueller, S.; Weng, H.; Dooley, S. Current experimental perspectives on the clinical progression of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2009, 33, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Pastorino, J.G. Cellular signaling mechanisms in alcohol-induced liver damage. Semin. Liver Dis. 2004, 24, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.E. Recent insights into the role of the innate immune system in the development of alcoholic liver disease. Exp. Biol. Med. (Maywood.) 2003, 228, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.J.; Dong, Q.; Bindas, J.; Piganelli, J.D.; Magill, A.; Reiser, J.; Kolls, J.K. TRIF and IRF-3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J. Immunol. 2008, 181, 3049–3056. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Nieto, N. Kupffer cells and alcoholic liver disease. Rev. Esp. Enferm. Dig. 2006, 98, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Cubero, F.J.; Urtasun, R.; Nieto, N. Alcohol and liver fibrosis. Semin. Liver Dis. 2009, 29, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; de Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, S.C.; Bucio, L.; Souza, V.; Hernandez, E.; Gonzalez, E.; Gomez-Quiroz, L.; Kershenobich, D.; Vargas-Vorackova, F.; Gutierrez-Ruiz, M.C. Effect of endotoxin pretreatment on hepatic stellate cell response to ethanol and acetaldehyde. J. Gastroenterol. Hepatol. 2001, 16, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Lian, L.H.; Wu, Y.L.; Wan, Y.; Nan, J.X. Thymoquinone attenuates liver fibrosis via PI3K and TLR4 signaling pathways in activated hepatic stellate cells. Int. Immunopharmacol. 2013, 15, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Novo, E.; Parola, M. Redox mechanisms in hepatic chronic wound healing and fibrogenesis. Fibrogenesis Tissue Repair 2008, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Kobayashi, S.; Amano, M.; Kimura, K.; Kanaide, H.; Nakano, T.; Kaibuchi, K.; Ito, M. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. J. Biol. Chem. 1997, 272, 12257–12260. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, Y. Endothelial dysfunction in the regulation of cirrhosis and portal hypertension. Liver Int. 2012, 32, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Jagavelu, K.; Routray, C.; Shergill, U.; O’Hara, S.P.; Faubion, W.; Shah, V.H. Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 2010, 52, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, B.R.; Rogers, G.W.; Xing, H.Y.; Fraser, R. Endotoxin-induced defenestration of the hepatic sinusoidal endothelium: A factor in the pathogenesis of cirrhosis? Liver 1994, 14, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef] [PubMed]
- Seth, D.; Haber, P.S.; Syn, W.K.; Diehl, A.M.; Day, C.P. Pathogenesis of alcohol-induced liver disease: Classical concepts and recent advances. J. Gastroenterol. Hepatol. 2011, 26, 1089–1105. [Google Scholar] [CrossRef] [PubMed]
- Zima, T.; Kalousova, M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 110S–115S. [Google Scholar] [CrossRef] [PubMed]
- Mezey, E.; Potter, J.J.; Rennie-Tankersley, L.; Caballeria, J.; Pares, A. A randomized placebo controlled trial of vitamin E for alcoholic hepatitis. J. Hepatol. 2004, 40, 40–46. [Google Scholar] [CrossRef]
- Stewart, S.; Prince, M.; Bassendine, M.; Hudson, M.; James, O.; Jones, D.; Record, C.; Day, C.P. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J. Hepatol. 2007, 47, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Langlet, P.; Hittelet, A.; Lasser, L.; Degre, D.; Evrard, S.; Colle, I.; Lemmers, A.; Deviere, J.; Le, M.O. Enteral nutrition with or without N-acetylcysteine in the treatment of severe acute alcoholic hepatitis: A randomized multicenter controlled trial. J. Hepatol. 2010, 53, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Dakin, H.D.; Dudley, H.W. An enzyme concerned with the formation of hydroxy acids from ketonic aldehydes. J. Biol. Chem. 1913, 14, 155–157. [Google Scholar]
- Weaver, R.H.; Lardy, H.A. Synthesis and some biochemical properties of phosphohydroxypyruvic aldehyde and of 3-phosphoglyceryl glutathione thiol ester. J. Biol. Chem. 1961, 236, 313–317. [Google Scholar] [PubMed]
- Vander Jagt, D.L. The glyoxalase system. In Glutathione “Chemical, Biochemical and Medical Aspects”; Wiley: New York, NY, USA, 1989; pp. 597–641. [Google Scholar]
- Mannervik, B. Glyoxalase I. Enzymatic Basis of Detoxification; Academic Press: New York, NY, USA, 1980; Volume 2, pp. 263–293. [Google Scholar]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Aspects Med. 1993, 14, 287–371. [Google Scholar] [CrossRef]
- Racker, E. The mechanism of action of glyoxalase. J. Biol. Chem. 1951, 190, 685–696. [Google Scholar] [PubMed]
- Hopkins, F.G.; Morgan, E.J. On the distribution of glyoxalase and glutathione. Biochem. J. 1945, 39, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [PubMed]
- Misra, K.; Banerjee, A.B.; Ray, S.; Ray, M. Glyoxalase III from Escherichia coli: A single novel enzyme for the conversion of methylglyoxal into D-lactate without reduced glutathione. Biochem. J. 1995, 305, 999–1003. [Google Scholar] [CrossRef] [PubMed]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, S.; Mori, M.; Shiraha, K.; Kawase, M. Biosynthesis and degradation of methylglyoxal in animals. Prog. Clin. Biol. Res. 1989, 290, 397–412. [Google Scholar] [PubMed]
- Ray, S.; Ray, M. Formation of methylglyoxal from aminoacetone by amine oxidase from goat plasma. J. Biol. Chem. 1983, 258, 3461–3462. [Google Scholar] [PubMed]
- Casazza, J.P.; Felver, M.E.; Veech, R.L. The metabolism of acetone in rat. J. Biol. Chem. 1984, 259, 231–236. [Google Scholar] [PubMed]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. Protein and nucleotide damage by glyoxal and methylglyoxal in physiological systems—Role in ageing and disease. Drug Metab. Drug Interact. 2008, 23, 125–150. [Google Scholar] [CrossRef]
- Vaca, C.E.; Fang, J.L.; Conradi, M.; Hou, S.M. Development of a 32P-postlabelling method for the analysis of 2′-deoxyguanosine-3′-monophosphate and DNA adducts of methylglyoxal. Carcinogenesis 1994, 15, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Westwood, M.E.; McLellan, A.C.; Selwood, T.; Thornalley, P.J. Binding and modification of proteins by methylglyoxal under physiological conditions. A kinetic and mechanistic study with N alpha-acetylarginine, N alpha-acetylcysteine, and N alpha-acetyllysine, and bovine serum albumin. J. Biol. Chem. 1994, 269, 32299–32305. [Google Scholar] [PubMed]
- Shipanova, I.N.; Glomb, M.A.; Nagaraj, R.H. Protein modification by methylglyoxal: Chemical nature and synthetic mechanism of a major fluorescent adduct. Arch. Biochem. Biophys. 1997, 344, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Oya, T.; Hattori, N.; Mizuno, Y.; Miyata, S.; Maeda, S.; Osawa, T.; Uchida, K. Methylglyoxal modification of protein. Chemical and immunochemical characterization of methylglyoxal-arginine adducts. J. Biol. Chem. 1999, 274, 18492–18502. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Munch, G.; Keis, R.; Wessels, A.; Riederer, P.; Bahner, U.; Heidland, A.; Niwa, T.; Lemke, H.D.; Schinzel, R. Determination of advanced glycation end products in serum by fluorescence spectroscopy and competitive ELISA. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, A.; Uchida, K.; Nakanishi, M.; Wakabayashi, I. Measurement of urinary advanced glycation end-products (AGEs) using a fluorescence assay for metabolic syndrome-related screening tests. Diabetes Metab. Syndr. 2016, 10, S110–S113. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 2012, 42, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of LDL by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Nass, N.; Sel, S.; Ignatov, A.; Roessner, A.; Kalinski, T. Oxidative stress and glyoxalase I activity mediate dicarbonyl toxicity in MCF-7 mamma carcinoma cells and a tamoxifen resistant derivative. Biochim. Biophys. Acta 2016, 1860, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Saito, K.; Sato, E.; Nakayama, K.; Terawaki, H.; Ito, S.; Kohno, M. Radical generation by the non-enzymatic reaction of methylglyoxal and hydrogen peroxide. Redox Rep. 2007, 12, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.T.; Merchant, M.L.; Kain, A.B.; Jevans, A.W.; McLeish, K.R.; Klein, J.B. Proteomic analysis defines altered cellular redox pathways and advanced glycation end-product metabolism in glomeruli of db/db diabetic mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F1157–F1165. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Beulens, J.W.; van Dieren, S.; Scheijen, J.L.; van der A, D.L.; Spijkerman, A.M.; van der Schouw, Y.T.; Stehouwer, C.D.; Schalkwijk, C.G. Plasma advanced glycation end products are associated with incident cardiovascular events in individuals with type 2 diabetes: A case-cohort study with a median follow-up of 10 years (EPIC-NL). Diabetes 2015, 64, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Agalou, S.; Ahmed, N.; Babaei-Jadidi, R.; Dawnay, A.; Thornalley, P.J. Profound mishandling of protein glycation degradation products in uremia and dialysis. J. Am. Soc. Nephrol. 2005, 16, 1471–1485. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. Biophys. Acta 2000, 1498, 99–111. [Google Scholar] [CrossRef]
- Piperi, C.; Goumenos, A.; Adamopoulos, C.; Papavassiliou, A.G. AGE/RAGE signalling regulation by miRNAs: Associations with diabetic complications and therapeutic potential. Int. J. Biochem. Cell Biol. 2015, 60, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Li, Q.; Li, M.; Wang, W.; Cui, C.; Zhang, J. Ghrelin inhibits AGEs-induced apoptosis in human endothelial cells involving ERK1/2 and PI3K/Akt pathways. Cell Biochem. Funct. 2011, 29, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; He, J.C.; Cai, W.; Liu, H.; Zhu, L.; Vlassara, H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 11767–11772. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, R.; Yan, S.F.; Schmidt, A.M. Advanced glycation endproducts: From precursors to RAGE: Round and round we go. Amino Acids 2012, 42, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Akhand, A.A.; Hossain, K.; Mitsui, H.; Kato, M.; Miyata, T.; Inagi, R.; Du, J.; Takeda, K.; Kawamoto, Y.; Suzuki, H.; et al. Glyoxal and methylglyoxal trigger distinct signals for map family kinases and caspase activation in human endothelial cells. Free Radic. Biol. Med. 2001, 31, 20–30. [Google Scholar] [CrossRef]
- Du, J.; Suzuki, H.; Nagase, F.; Akhand, A.A.; Yokoyama, T.; Miyata, T.; Kurokawa, K.; Nakashima, I. Methylglyoxal induces apoptosis in Jurkat leukemia T cells by activating c-Jun N-terminal kinase. J. Cell. Biochem. 2000, 77, 333–344. [Google Scholar] [CrossRef]
- Miglietta, A.; Gabriel, L. Methylglyoxal-tubulin interaction: Studies on the aldehyde effects on hepatoma, liver and purified microtubular protein. Res. Commun. Chem. Pathol. Pharmacol. 1986, 51, 245–260. [Google Scholar] [PubMed]
- Wu, L.; Juurlink, B.H. Increased methylglyoxal and oxidative stress in hypertensive rat vascular smooth muscle cells. Hypertension 2002, 39, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Han, L.P.; Schimandle, C.M.; Davison, L.M.; Vander Jagt, D.L. Comparative kinetics of Mg2+-, Mn2+-, Co2+-, and Ni2+-activated glyoxalase I. Evaluation of the role of the metal ion. Biochemistry 1977, 16, 5478–5484. [Google Scholar] [CrossRef] [PubMed]
- Sellin, S.; Eriksson, L.E.; Aronsson, A.C.; Mannervik, B. Octahedral metal coordination in the active site of glyoxalase I as evidenced by the properties of Co(II)-glyoxalase I. J. Biol. Chem. 1983, 258, 2091–2093. [Google Scholar] [PubMed]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.D.; Olin, B.; Ridderstrom, M.; Mannervik, B.; Jones, T.A. Crystal structure of human glyoxalase I—Evidence for gene duplication and 3D domain swapping. EMBO J. 1997, 16, 3386–3395. [Google Scholar] [CrossRef] [PubMed]
- Kompf, J.; Bissbort, S.; Gussmann, S.; Ritter, H. Polymorphism of red cell glyoxalase I (EI: 4.4.1.5); a new genetic marker in man. Investigation of 169 mother-child combinations. Humangenetik 1975, 27, 141–143. [Google Scholar] [PubMed]
- Kompf, J.; Bissbort, S.; Ritter, H. Red cell glyoxalase i (E.C.: 4.4.1.5): Formal genetics and linkage relations. Humangenetik 1975, 28, 249–251. [Google Scholar] [PubMed]
- Thornalley, P.J. Population genetics of human glyoxalases. Heredity 1991, 67, 139–142. [Google Scholar] [CrossRef] [PubMed]
- McCann, V.J.; Davis, R.E.; Welborn, T.A. Glyoxalase phenotypes in patients with diabetes mellitus. Aust. N. Z. J. Med. 1981, 11, 380–382. [Google Scholar] [CrossRef] [PubMed]
- Gale, C.P.; Futers, T.S.; Summers, L.K. Common polymorphisms in the glyoxalase-1 gene and their association with pro-thrombotic factors. Diabetes Vasc. Dis. Res. 2004, 1, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Bangel, F.N.; Yamada, K.; Arai, M.; Iwayama, Y.; Balan, S.; Toyota, T.; Iwata, Y.; Suzuki, K.; Kikuchi, M.; Hashimoto, T.; et al. Genetic analysis of the glyoxalase system in schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 59, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Barua, M.; Jenkins, E.C.; Chen, W.; Kuizon, S.; Pullarkat, R.K.; Junaid, M.A. Glyoxalase I polymorphism rs2736654 causing the Ala111Glu substitution modulates enzyme activity--implications for autism. Autism Res. 2011, 4, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Junaid, M.A.; Kowal, D.; Barua, M.; Pullarkat, P.S.; Sklower, B.S.; Pullarkat, R.K. Proteomic studies identified a single nucleotide polymorphism in glyoxalase I as autism susceptibility factor. Am. J. Med. Genet. A 2004, 131, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Sidoti, A.; Antognelli, C.; Rinaldi, C.; D'Angelo, R.; Dattola, V.; Girlanda, P.; Talesa, V.; Amato, A. Glyoxalase I A111E, paraoxonase 1 Q192R and L55M polymorphisms: Susceptibility factors of multiple sclerosis? Mult. Scler. 2007, 13, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.; Lim, J.E.; Harr, B.; Wing, C.; Walters, R.; Distler, M.G.; Teschke, M.; Wu, C.; Wiltshire, T.; Su, A.I.; et al. A common and unstable copy number variant is associated with differences in Glo1 expression and anxiety-like behavior. PLoS ONE 2009, 4, e4649. [Google Scholar] [CrossRef] [PubMed]
- Santarius, T.; Bignell, G.R.; Greenman, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. GLO1-A novel amplified gene in human cancer. Genes Chromosom. Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Shafie, A.; Xue, M.; Thornalley, P.J.; Rabbani, N. Copy number variation of glyoxalase I. Biochem. Soc. Trans. 2014, 42, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Mezzasoma, L.; Mearini, E.; Talesa, V.N. Glyoxalase 1-419C > A variant is associated with oxidative stress: Implications in prostate cancer progression. PLoS ONE 2013, 8, e74014. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Tisdale, M.J. Inhibition of proliferation of human promyelocytic leukaemia HL60 cells by S-d-lactoylglutathione in vitro. Leuk. Res. 1988, 12, 897–904. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Edwards, L.G.; Kang, Y.; Wyatt, C.; Davies, N.; Ladan, M.J.; Double, J. Antitumour activity of S-p-bromobenzylglutathione cyclopentyl diester in vitro and in vivo. Inhibition of glyoxalase I and induction of apoptosis. Biochem. Pharmacol. 1996, 51, 1365–1372. [Google Scholar] [CrossRef]
- Baunacke, M.; Horn, L.C.; Trettner, S.; Engel, K.M.; Hemdan, N.Y.; Wiechmann, V.; Stolzenburg, J.U.; Bigl, M.; Birkenmeier, G. Exploring glyoxalase 1 expression in prostate cancer tissues: Targeting the enzyme by ethyl pyruvate defangs some malignancy-associated properties. Prostate 2014, 74, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Birkenmeier, G.; Hemdan, N.Y.; Kurz, S.; Bigl, M.; Pieroh, P.; Debebe, T.; Buchold, M.; Thieme, R.; Wichmann, G.; Dehghani, F. Ethyl Pyruvate Combats Human Leukemia Cells but Spares Normal Blood Cells. PLoS ONE 2016, 11, e0161571. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [PubMed]
- Lo, T.W.; Thornalley, P.J. Inhibition of proliferation of human leukaemia 60 cells by diethyl esters of glyoxalase inhibitors in vitro. Biochem. Pharmacol. 1992, 44, 2357–2363. [Google Scholar] [CrossRef]
- Bartyik, K.; Turi, S.; Orosz, F.; Karg, E. Methotrexate inhibits the glyoxalase system in vivo in children with acute lymphoid leukaemia. Eur. J. Cancer 2004, 40, 2287–2292. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Kwon, Y.; Kamisuki, S.; Srivastava, N.; Mao, Q.; Kawazoe, Y.; Uesugi, M. Polyproline-rod approach to isolating protein targets of bioactive small molecules: Isolation of a new target of indomethacin. J. Am. Chem. Soc. 2007, 129, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Eftekharpour, E.; Davies, G.F.; Roesler, W.J.; Juurlink, B.H. Troglitazone selectively inhibits glyoxalase I gene expression. Diabetologia 2001, 44, 2004–2012. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, R.; Takahashi, S.; Saeki, K.; Sunaga, S.; Yoshimori, A.; Tanuma, S.I. Structure-activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg. Med. Chem. 2008, 16, 3969–3975. [Google Scholar] [CrossRef] [PubMed]
- Santel, T.; Pflug, G.; Hemdan, N.Y.; Schafer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M.; Thonig, A.; Pohl, S.; Ripoll, C.; Michel, M.; Zipprich, A. Expression of glyoxalase-I is reduced in cirrhotic livers: A possible mechanism in the development of cirrhosis. PLoS ONE 2017, 12, e0171260. [Google Scholar] [CrossRef] [PubMed]
- Hollenbach, M.; Hintersdorf, A.; Huse, K.; Sack, U.; Bigl, M.; Groth, M.; Santel, T.; Buchold, M.; Lindner, I.; Otto, A.; et al. Ethyl pyruvate and ethyl lactate down-regulate the production of pro-inflammatory cytokines and modulate expression of immune receptors. Biochem. Pharmacol. 2008, 76, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.P. Ethyl pyruvate: A novel treatment for sepsis. Novartis Found. Symp. 2007, 280, 147–156. [Google Scholar] [PubMed]
- Sims, C.A.; Wattanasirichaigoon, S.; Menconi, M.J.; Ajami, A.M.; Fink, M.P. Ringer’s ethyl pyruvate solution ameliorates ischemia/reperfusion-induced intestinal mucosal injury in rats. Crit. Care Med. 2001, 29, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Delude, R.L.; Fink, M.P. Dose-dependent effects of ethyl pyruvate in mice subjected to mesenteric ischemia and reperfusion. Intensiv. Care Med. 2003, 29, 2050–2058. [Google Scholar] [CrossRef] [PubMed]
- Cruz, R.J., Jr.; Harada, T.; Sasatomi, E.; Fink, M.P. Effects of ethyl pyruvate and other alpha-keto carboxylic acid derivatives in a rat model of multivisceral ischemia and reperfusion. J. Surg. Res. 2011, 165, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gallo, D.J.; Baust, J.J.; Uchiyama, T.; Watkins, S.K.; Delude, R.L.; Fink, M.P. Ethyl pyruvate modulates inflammatory gene expression in mice subjected to hemorrhagic shock. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G212–G221. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, R.; Kellum, J.A.; Baust, J.J.; Uchiyama, T.; Watkins, S.K.; Delude, R.L.; Fink, M.P. Resuscitation with Ringer’s ethyl pyruvate solution prolongs survival and modulates plasma cytokine and nitrite/nitrate concentrations in a rat model of lipopolysaccharide-induced shock. Shock 2002, 18, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Hauser, B.; Kick, J.; Asfar, P.; Ehrmann, U.; Albicini, M.; Vogt, J.; Wachter, U.; Bruckner, U.B.; Fink, M.P.; Radermacher, P.; et al. Ethyl pyruvate improves systemic and hepatosplanchnic hemodynamics and prevents lipid peroxidation in a porcine model of resuscitated hyperdynamic endotoxemia. Crit. Care Med. 2005, 33, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Ochani, M.; Yang, H.; Tanovic, M.; Halperin, D.; Yang, R.; Czura, C.J.; Fink, M.P.; Tracey, K.J. Ethyl pyruvate prevents lethality in mice with established lethal sepsis and systemic inflammation. Proc. Natl. Acad. Sci. USA 2002, 99, 12351–12356. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, T.; Hu, X.; Yuen, P.S.; Muramatsu, Y.; Iyer, S.; Hewitt, S.M.; Star, R.A. Ethyl pyruvate decreases sepsis-induced acute renal failure and multiple organ damage in aged mice. Kidney Int. 2003, 64, 1620–1631. [Google Scholar] [CrossRef] [PubMed]
- Reade, M.C.; Fink, M.P. Bench-to-bedside review: Amelioration of acute renal impairment using ethyl pyruvate. Crit. Care 2005, 9, 556–560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.Q.; Liu, C.T.; Li, W.J.; Fan, W.; Zhong, N.; Zhang, Y.; Jia, X.Q.; Zhang, S.Z. Ethyl pyruvate improves survival and ameliorates distant organ injury in rats with severe acute pancreatitis. Pancreas 2007, 35, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.G.; Zhang, H.; Ma, X.C.; Zhang, C.; Guo, R.X. Therapeutic treatment with ethyl pyruvate attenuates the severity of liver injury in rats with severe acute pancreatitis. Pancreas 2012, 41, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Luan, Z.G.; Ma, X.C.; Zhang, H.; Zhang, C.; Guo, R.X. Protective effect of ethyl pyruvate on pancreas injury in rats with severe acute pancreatitis. J. Surg. Res. 2013, 181, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Uchiyama, T.; Alber, S.M.; Han, X.; Watkins, S.K.; Delude, R.L.; Fink, M.P. Ethyl pyruvate ameliorates distant organ injury in a murine model of acute necrotizing pancreatitis. Crit. Care Med. 2004, 32, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Karabeyoglu, M.; Unal, B.; Bozkurt, B.; Dolapci, I.; Bilgihan, A.; Karabeyoglu, I.; Cengiz, O. The effect of ethyl pyruvate on oxidative stress in intestine and bacterial translocation after thermal injury. J. Surg. Res. 2008, 144, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Jeong, J.Y.; Kim, H.J.; Seo, J.S.; Han, P.L.; Yoon, S.H.; Lee, J.K. Combination treatment with ethyl pyruvate and aspirin enhances neuroprotection in the postischemic brain. Neurotox. Res. 2010, 17, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kim, H.S. Inhibitory mechanism of MMP-9 gene expression by ethyl pyruvate in lipopolysaccharide-stimulated BV2 microglial cells. Neurosci. Lett. 2011, 493, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Moro, N.; Sutton, R.L. Beneficial effects of sodium or ethyl pyruvate after traumatic brain injury in the rat. Exp. Neurol. 2010, 225, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Hu, X.; Liu, C.; Wang, S.; Zhang, W.; Gao, H.; Stetler, R.A.; Gao, Y.; Chen, J. Ethyl pyruvate protects against hypoxic-ischemic brain injury via anti-cell death and anti-inflammatory mechanisms. Neurobiol. Dis. 2010, 37, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wang, H.; Zhu, L.; Zhao, J.; Pan, H.; Ji, X. Ethyl pyruvate ameliorates intracerebral hemorrhage-induced brain injury through anti-cell death and anti-inflammatory mechanisms. Neuroscience 2013, 245, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Jang, I.S.; Park, M.Y.; Shin, I.W.; Sohn, J.T.; Lee, H.K.; Chung, Y.K. Ethyl pyruvate has anti-inflammatory and delayed myocardial protective effects after regional ischemia/reperfusion injury. Yonsei Med. J. 2010, 51, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Woo, Y.J.; Taylor, M.D.; Cohen, J.E.; Jayasankar, V.; Bish, L.T.; Burdick, J.; Pirolli, T.J.; Berry, M.F.; Hsu, V.; Grand, T. Ethyl pyruvate preserves cardiac function and attenuates oxidative injury after prolonged myocardial ischemia. J. Thorac. Cardiovasc. Surg. 2004, 127, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, Y.M.; Kim, C.S.; Sohn, E.; Jo, K.; Shin, S.D.; Kim, J.S. Ethyl pyruvate prevents methyglyoxal-induced retinal vascular injury in rats. J. Diabetes Res. 2013, 2013, 460820. [Google Scholar] [CrossRef] [PubMed]
- Kalariya, N.M.; Reddy, A.B.; Ansari, N.H.; VanKuijk, F.J.; Ramana, K.V. Preventive effects of ethyl pyruvate on endotoxin-induced uveitis in rats. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5144–5152. [Google Scholar] [CrossRef] [PubMed]
- Devamanoharan, P.S.; Henein, M.; Ali, A.H.; Varma, S.D. Attenuation of sugar cataract by ethyl pyruvate. Mol. Cell. Biochem. 1999, 200, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Mohanty, I.; Trivedi, D.; Tandon, R.; Srivastava, S.; Joshi, S. Pyruvate inhibits galactosemic changes in cultured cat lens epithelial cells. Ophthalmic Res. 2002, 34, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.D.; Hegde, K.R.; Kovtun, S. Oxidative damage to lens in culture: Reversibility by pyruvate and ethyl pyruvate. Ophthalmologica 2006, 220, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Devamanoharan, P.S.; Henein, M.; Ali, A.H.; Varma, S.D. Diabetes-induced biochemical changes in rat lens: Attenuation of cataractogenesis by pyruvate. Diabetes Obes. Metab. 2000, 2, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, J.S.; Zhou, Z.; Chen, W.X.; Chen, N.W. Inhibitory effects of ethyl pyruvate administration on human gastric cancer growth via regulation of the HMGB1-RAGE and Akt pathways in vitro and in vivo. Oncol. Rep. 2012, 27, 1511–1519. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Wang, X.F.; Tan, Z.J.; Dong, P.; Gu, J.; Lu, J.H.; Wu, X.S.; Zhang, L.; Ding, Q.C.; Wu, W.G.; et al. Ethyl pyruvate administration suppresses growth and invasion of gallbladder cancer cells via downregulation of HMGB1-RAGE axis. Int. J. Immunopathol. Pharmacol. 2012, 25, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Wang, L.K.; Chen, H.; Fan, C.; Li, X.; He, C.M.; Gong, Z.J. Ethyl pyruvate protects against experimental acute-on-chronic liver failure in rats. World J. Gastroenterol. 2012, 18, 5709–5718. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Han, X.; Delude, R.L.; Fink, M.P. Ethyl pyruvate ameliorates acute alcohol-induced liver injury and inflammation in mice. J. Lab. Clin. Med. 2003, 142, 322–331. [Google Scholar] [CrossRef]
- Yang, R.; Shaufl, A.L.; Killeen, M.E.; Fink, M.P. Ethyl pyruvate ameliorates liver injury secondary to severe acute pancreatitis. J. Surg. Res. 2009, 153, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zou, X.; Koskinen, M.L.; Tenhunen, J. Ethyl pyruvate reduces liver injury at early phase but impairs regeneration at late phase in acetaminophen overdose. Crit. Care 2012, 16, R9. [Google Scholar] [CrossRef] [PubMed]
- Olek, R.A.; Ziolkowski, W.; Flis, D.J.; Fedeli, D.; Fiorini, D.; Wierzba, T.H.; Gabbianelli, R. The effect of ethyl pyruvate supplementation on rat fatty liver induced by a high-fat diet. J. Nutr. Sci. Vitaminol. (Tokyo) 2013, 59, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Bennett-Guerrero, E.; Swaminathan, M.; Grigore, A.M.; Roach, G.W.; Aberle, L.G.; Johnston, J.M.; Fink, M.P. A phase II multicenter double-blind placebo-controlled study of ethyl pyruvate in high-risk patients undergoing cardiac surgery with cardiopulmonary bypass. J. Cardiothorac. Vasc. Anesth. 2009, 23, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Thornalley, P.J.; Grigore, A.M.; Roach, G.W.; Aberle, L.G.; Johnston, J.M.; Fink, M.P. Processing of protein glycation, oxidation and nitrosation adducts in the liver and the effect of cirrhosis. J. Hepatol. 2004, 41, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Sebekova, K.; Kupcova, V.; Schinzel, R.; Heidland, A. Markedly elevated levels of plasma advanced glycation end products in patients with liver cirrhosis—Amelioration by liver transplantation. J. Hepatol. 2002, 36, 66–71. [Google Scholar] [CrossRef]
- Yagmur, E.; Tacke, F.; Weiss, C.; Lahme, B.; Manns, M.P.; Kiefer, P.; Trautwein, C.; Gressner, A.M. Elevation of Nepsilon-(carboxymethyl)lysine-modified advanced glycation end products in chronic liver disease is an indicator of liver cirrhosis. Clin. Biochem. 2006, 39, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Zuwala-Jagiello, J.; Pazgan-Simon, M.; Simon, K.; Warwas, M. Elevated advanced oxidation protein products levels in patients with liver cirrhosis. Acta Biochim. Pol. 2009, 56, 679–685. [Google Scholar] [PubMed]
- Guimaraes, E.L.; Empsen, C.; Geerts, A.; van Grunsven, L.A. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J. Hepatol. 2010, 52, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, R.; Andrades, M.E.; Bortolin, R.C.; Nagai, R.; Dal-Pizzol, F.; Moreira, J.C. Oxidative damage in the liver of rats treated with glycolaldehyde. Int. J. Toxicol. 2011, 30, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Kanno, K.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Tazuma, S.; Chayama, K. Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J. Gastroenterol. 2008, 43, 298–304. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; George, J.; Takeuchi, M.; Fukumura, A.; Toshikuni, N.; Arisawa, T.; Tsutsumi, M. Acetaldehyde-derived advanced glycation end-products promote alcoholic liver disease. PLoS ONE 2013, 8, e70034. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhu, J.; Huang, Y.; Gao, H.; Zhao, Y. Advanced glycation end product (AGE)-induced hepatic stellate cell activation via autophagy contributes to hepatitis C-related fibrosis. Acta Diabetol. 2015, 52, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.H.; Lee, B.H.; Hsu, Y.W.; Pan, T.M. Peroxisome proliferator-activated receptor-gamma activators monascin and rosiglitazone attenuate carboxymethyllysine-induced fibrosis in hepatic stellate cells through regulating the oxidative stress pathway but independent of the receptor for advanced glycation end products signaling. J. Agric. Food Chem. 2013, 61, 6873–6879. [Google Scholar] [PubMed]
- Goodwin, M.; Herath, C.; Jia, Z.; Leung, C.; Coughlan, M.T.; Forbes, J.; Angus, P. Advanced glycation end products augment experimental hepatic fibrosis. J. Gastroenterol. Hepatol. 2013, 28, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nepsilon-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Vlassara, H. Recent progress in advanced glycation end products and diabetic complications. Diabetes 1997, 46 (Suppl. S2), S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Baker, S.S.; Liu, W.; Desai, S.; Alkhouri, R.; Kozielski, R.; Mastrandrea, L.; Sarfraz, A.; Cai, W.; Vlassara, H.; et al. Effect of dietary advanced glycation end products on mouse liver. PLoS ONE 2012, 7, e35143. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Herath, C.B.; Jia, Z.; Goodwin, M.; Mak, K.Y.; Watt, M.J.; Forbes, J.M.; Angus, P.W. Dietary glycotoxins exacerbate progression of experimental fatty liver disease. J. Hepatol. 2014, 60, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Chen, X.; Fukada, H.; Serizawa, N.; Devaraj, S.; Torok, N.J. Advanced glycation endproducts induce fibrogenic activity in nonalcoholic steatohepatitis by modulating TNF-alpha-converting enzyme activity in mice. Hepatology 2013, 58, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, A. Curcumin eliminates the effect of advanced glycation end-products (AGEs) on the divergent regulation of gene expression of receptors of AGEs by interrupting leptin signaling. Lab. Investig. 2014, 94, 503–516. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tang, Y.; Kang, Q.; Chen, A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab. Investig. 2012, 92, 827–841. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Hyogo, H.; Yamagishi, S.; Takeuchi, M.; Ishitobi, T.; Nabeshima, Y.; Arihiro, K.; Chayama, K. Atorvastatin decreases serum levels of advanced glycation endproducts (AGEs) in nonalcoholic steatohepatitis (NASH) patients with dyslipidemia: Clinical usefulness of AGEs as a biomarker for the attenuation of NASH. J. Gastroenterol. 2010, 45, 750–757. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kitahara, Y.; Yamagishi, S. Combination therapy with nateglinide and vildagliptin improves postprandial metabolic derangements in Zucker fatty rats. Horm. Metab. Res. 2010, 42, 731–735. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.J.; Choong, C.Y.; Shi, Y.C.; Lin, Y.C.; Wang, C.W.; Lee, B.H.; Tai, C.J. Solanum nigrum Protects against Hepatic Fibrosis via Suppression of Hyperglycemia in High-Fat/Ethanol Diet-Induced Rats. Molecules 2016, 21, 269. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.X.; Requena, J.R.; Jenkins, A.J.; Lyons, T.J.; Baynes, J.W.; Thorpe, S.R. The advanced glycation end product, Nepsilon-(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J. Biol. Chem. 1996, 271, 9982–9986. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Barbezier, N.; Tessier, F.J.; Chango, A. [Receptor of advanced glycation endproducts RAGE/AGER: An integrative view for clinical applications]. Ann. Biol. Clin. (Paris) 2014, 72, 669–680. [Google Scholar] [PubMed]
- Fehrenbach, H.; Weiskirchen, R.; Kasper, M.; Gressner, A.M. Up-regulated expression of the receptor for advanced glycation end products in cultured rat hepatic stellate cells during transdifferentiation to myofibroblasts. Hepatology 2001, 34, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Lohwasser, C.; Neureiter, D.; Popov, Y.; Bauer, M.; Schuppan, D. Role of the receptor for advanced glycation end products in hepatic fibrosis. World J. Gastroenterol. 2009, 15, 5789–5798. [Google Scholar] [CrossRef] [PubMed]
- Serban, A.I.; Stanca, L.; Geicu, O.I.; Munteanu, M.C.; Dinischiotu, A. RAGE and TGF-beta1 Cross-Talk Regulate Extracellular Matrix Turnover and Cytokine Synthesis in AGEs Exposed Fibroblast Cells. PLoS ONE 2016, 11, e0152376. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Tang, Y.; Kang, Q.; Feng, Y.; Chen, A. Curcumin inhibits gene expression of receptor for advanced glycation end-products (RAGE) in hepatic stellate cells in vitro by elevating PPARgamma activity and attenuating oxidative stress. Br. J. Pharmacol. 2012, 166, 2212–2227. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.R.; Chen, T.T.; Li, W.D.; Lu, F.L.; Liu, J.; Cai, X.G.; Lu, Q.; Yang, C.P. Inhibitory effect of receptor for advanced glycation end productspecific small interfering RNAs on the development of hepatic fibrosis in primary rat hepatic stellate cells. Mol. Med. Rep. 2015, 12, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.G.; Xia, J.R.; Li, W.D.; Lu, F.L.; Liu, J.; Lu, Q.; Zhi, H. Anti-fibrotic effects of specific-siRNA targeting of the receptor for advanced glycation end products in a rat model of experimental hepatic fibrosis. Mol. Med. Rep. 2014, 10, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Deng, Q.; Gao, J.; Yu, X.; Zhang, Y.; Li, J.; Guan, W.; Hu, J.; Tan, Q.; Zhou, L.; et al. Therapeutic effects of antigen affinity-purified polyclonal anti-receptor of advanced glycation end-product (RAGE) antibodies on cholestasis-induced liver injury in rats. Eur. J. Pharmacol. 2016, 779, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Lin, Y.C.; Tsai, M.S.; Sun, C.K.; Yuan, S.S.; Chang, C.Y.; Jawan, B.; Lee, P.H. Involvement of the nuclear high mobility group B1 peptides released from injured hepatocytes in murine hepatic fibrogenesis. Biochim. Biophys. Acta 2014, 1842, 1720–1732. [Google Scholar] [CrossRef] [PubMed]
- Kuhla, A.; Trieglaff, C.; Vollmar, B. Role of age and uncoupling protein-2 in oxidative stress, RAGE/AGE interaction and inflammatory liver injury. Exp. Gerontol. 2011, 46, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- Fattovich, G.; Giustina, G.; Degos, F.; Tremolada, F.; Diodati, G.; Almasio, P.; Nevens, F.; Solinas, A.; Mura, D.; Brouwer, J.T.; et al. Morbidity and mortality in compensated cirrhosis type C: A retrospective follow-up study of 384 patients. Gastroenterology 1997, 112, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Hicklin, D.J.; Huber, J.; Nakatani, T.; Tsujinoue, H.; Yanase, K.; et al. Synergistic effect of basic fibroblast growth factor and vascular endothelial growth factor in murine hepatocellular carcinoma. Hepatology 2002, 35, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Sugimachi, K.; Tanaka, S.; Kameyama, T.; Taguchi, K.; Aishima, S.; Shimada, M.; Sugimachi, K.; Tsuneyoshi, M. Transcriptional repressor snail and progression of human hepatocellular carcinoma. Clin. Cancer Res. 2003, 9, 2657–2664. [Google Scholar] [PubMed]
- Semela, D.; Dufour, J.F. Angiogenesis and hepatocellular carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef] [PubMed]
- Mise, M.; Arii, S.; Higashituji, H.; Furutani, M.; Niwano, M.; Harada, T.; Ishigami, S.; Toda, Y.; Nakayama, H.; Fukumoto, M.; et al. Clinical significance of vascular endothelial growth factor and basic fibroblast growth factor gene expression in liver tumor. Hepatology 1996, 23, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Yang, X.; He, Q.; Chen, Q.; Yu, L. Glyoxalase 1 is up-regulated in hepatocellular carcinoma and is essential for HCC cell proliferation. Biotechnol. Lett. 2014, 36, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liang, X.; Zheng, X.; Huang, H.; Chen, X.; Wu, K.; Wang, B.; Ma, S. Glo1 genetic amplification as a potential therapeutic target in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2014, 7, 2079–2090. [Google Scholar] [PubMed]
- Kan, H.; Yamagishi, S.; Ojima, A.; Fukami, K.; Ueda, S.; Takeuchi, M.; Hyogo, H.; Aikata, H.; Chayama, K. Elevation of Serum Levels of Advanced Glycation End Products in Patients With Non-B or Non-C Hepatocellular Carcinoma. J. Clin. Lab. Anal. 2015, 29, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Kohles, N.; Nagel, D.; Jungst, D.; Stieber, P.; Holdenrieder, S. Predictive value of immunogenic cell death biomarkers HMGB1, sRAGE, and DNase in liver cancer patients receiving transarterial chemoembolization therapy. Tumour Biol. 2012, 33, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Yaser, A.M.; Huang, Y.; Zhou, R.R.; Hu, G.S.; Xiao, M.F.; Huang, Z.B.; Duan, C.J.; Tian, W.; Tang, D.L.; Fan, X.G. The Role of receptor for Advanced Glycation End Products (RAGE) in the proliferation of hepatocellular carcinoma. Int. J. Mol. Sci. 2012, 13, 5982–5997. [Google Scholar] [CrossRef] [PubMed]
- Hiwatashi, K.; Ueno, S.; Abeyama, K.; Kubo, F.; Sakoda, M.; Maruyama, I.; Hamanoue, M.; Natsugoe, S.; Aikou, T. A novel function of the receptor for advanced glycation end-products (RAGE) in association with tumorigenesis and tumor differentiation of HCC. Ann. Surg. Oncol. 2008, 15, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Takino, J.; Yamagishi, S.; Takeuchi, M. Glycer-AGEs-RAGE signaling enhances the angiogenic potential of hepatocellular carcinoma by upregulating VEGF expression. World J. Gastroenterol. 2012, 18, 1781–1788. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhao, L.H.; Huang, B.; Wang, R.Y.; Yuan, S.X.; Tao, Q.F.; Xu, Y.; Sun, H.Y.; Lin, C.; Zhou, W.P. Pioglitazone, a PPARgamma agonist, inhibits growth and invasion of human hepatocellular carcinoma via blockade of the rage signaling. Mol. Carcinog. 2015, 54, 1584–1595. [Google Scholar] [CrossRef] [PubMed]
- Sakuraoka, Y.; Sawada, T.; Okada, T.; Shiraki, T.; Miura, Y.; Hiraishi, K.; Ohsawa, T.; Adachi, M.; Takino, J.; Takeuchi, M.; et al. MK615 decreases RAGE expression and inhibits TAGE-induced proliferation in hepatocellular carcinoma cells. World J. Gastroenterol. 2010, 16, 5334–5341. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Chang, Y.; Liang, X.; Cardinal, J.S.; Huang, H.; Thorne, S.H.; Monga, S.P.; Geller, D.A.; Lotze, M.T.; Tsung, A. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology 2012, 55, 1863–1875. [Google Scholar] [CrossRef] [PubMed]
- Moy, K.A.; Jiao, L.; Freedman, N.D.; Weinstein, S.J.; Sinha, R.; Virtamo, J.; Albanes, D.; Stolzenberg-Solomon, R.Z. Soluble receptor for advanced glycation end products and risk of liver cancer. Hepatology 2013, 57, 2338–2345. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hollenbach, M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). Int. J. Mol. Sci. 2017, 18, 2466. https://doi.org/10.3390/ijms18112466
Hollenbach M. The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). International Journal of Molecular Sciences. 2017; 18(11):2466. https://doi.org/10.3390/ijms18112466
Chicago/Turabian StyleHollenbach, Marcus. 2017. "The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC)" International Journal of Molecular Sciences 18, no. 11: 2466. https://doi.org/10.3390/ijms18112466
APA StyleHollenbach, M. (2017). The Role of Glyoxalase-I (Glo-I), Advanced Glycation Endproducts (AGEs), and Their Receptor (RAGE) in Chronic Liver Disease and Hepatocellular Carcinoma (HCC). International Journal of Molecular Sciences, 18(11), 2466. https://doi.org/10.3390/ijms18112466