Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature

Abstract
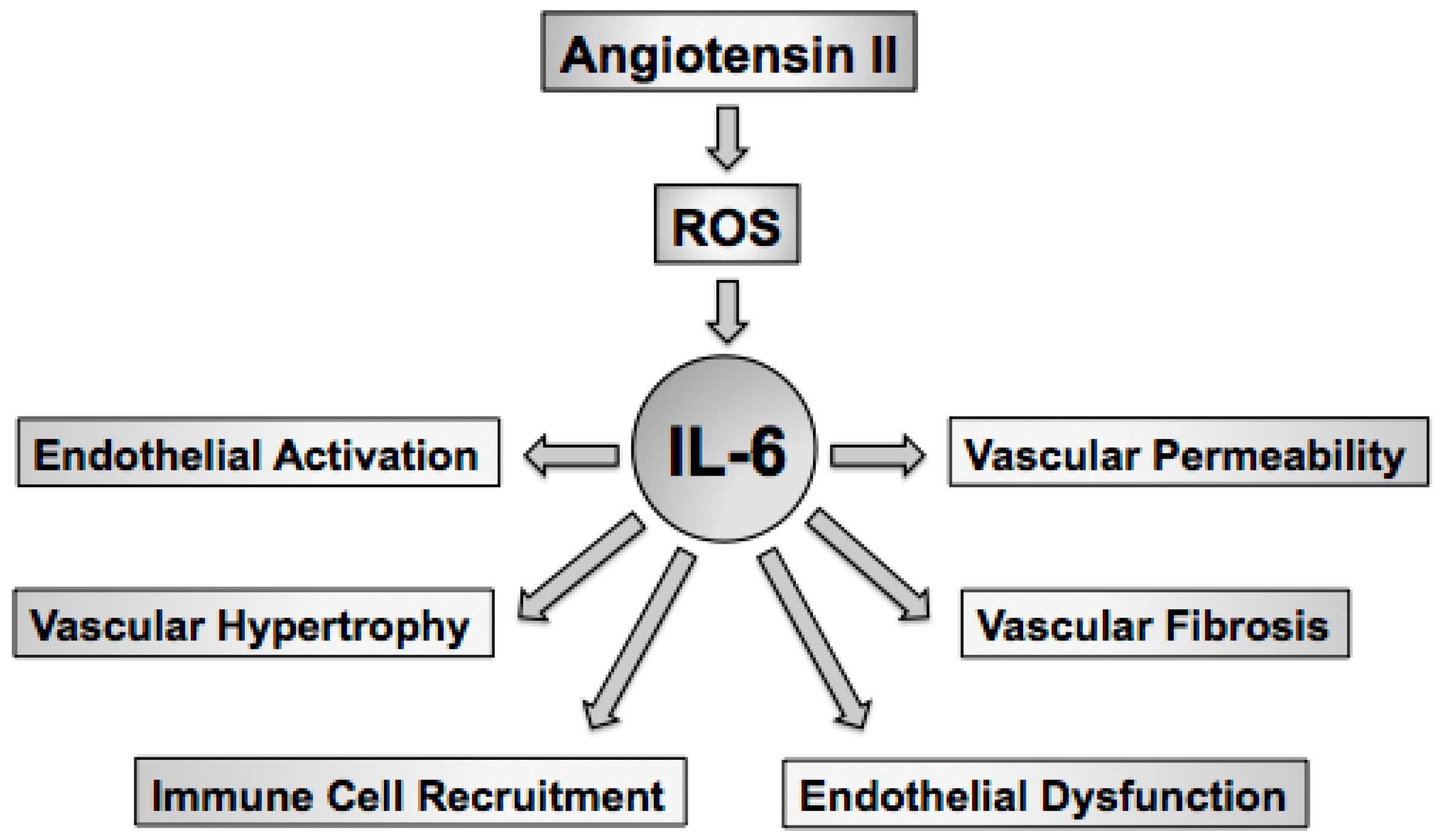
:1. Linking Oxidative Stress with Hypertension and Vascular Dysfunction
2. Interleukin-6
3. Interleukin-6 Signaling
4. Vascular Sources of Interleukin-6
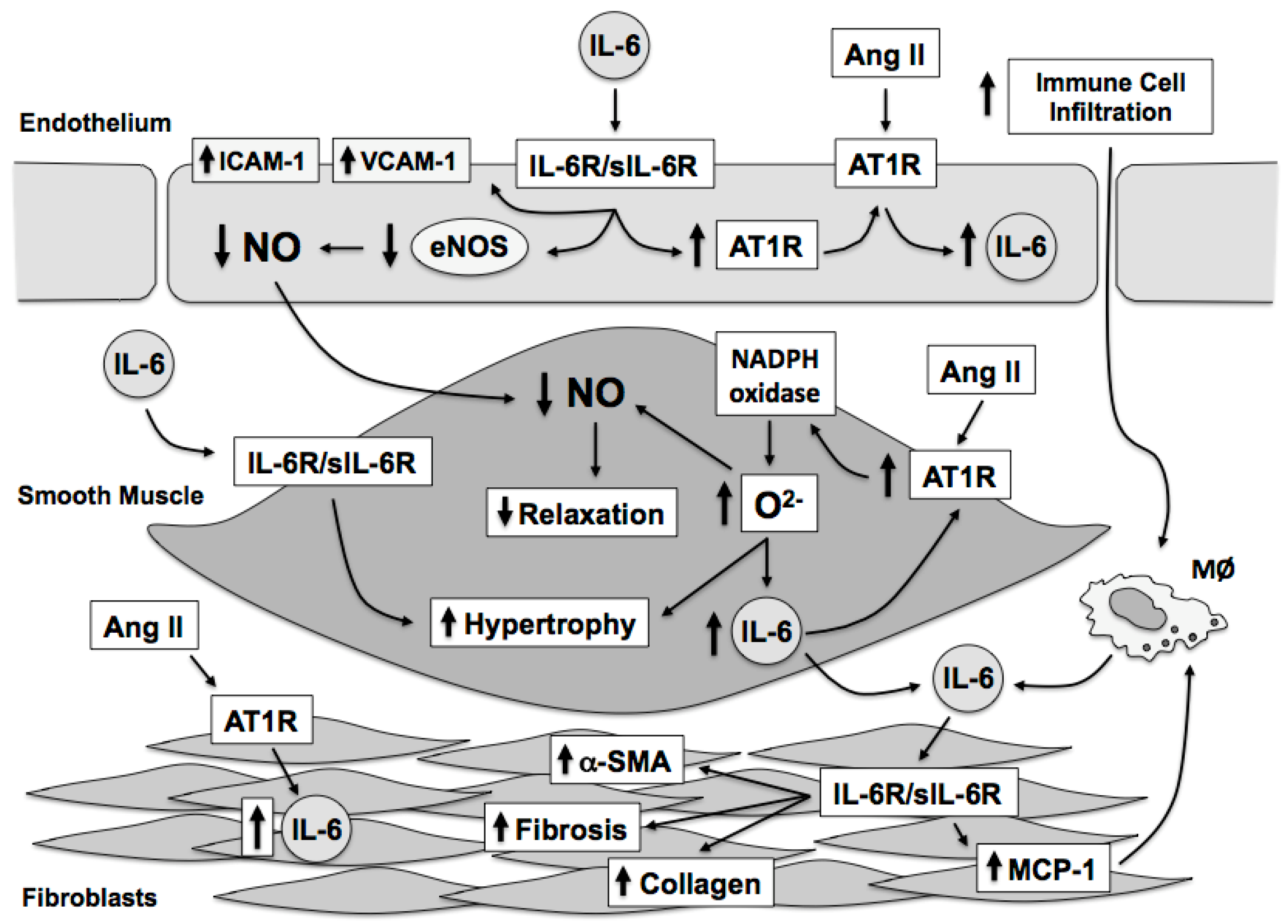
5. Effect of Interleukin-6 on Endothelial Cells
6. Effect of Interleukin-6 on Vascular Smooth Muscle
7. Effect of Interleukin-6 on Fibroblasts
8. Effects of Interleukin-6 on Endothelial Function and Oxidative Stress
9. Interleukin-6 and Blood Pressure
10. Summary
Acknowledgments
Conflicts of Interest
References
- Handy, D.E.; Loscalzo, J. Responses to reductive stress in the cardiovascular system. Free Radic. Biol. Med. 2017, 109, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Zhao, Y.; Xu, A.; Leung, S.W. Thirty years of saying NO: Sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circ. Res. 2016, 119, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J.; Touyz, R.M. Oxidative stress, inflammation, and vascular aging in hypertension. Hypertension 2017, 70, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Félétou, M.; Köhler, R.; Vanhoutte, P.M. Endothelium-derived vasoactive factors and hypertension: Possible roles in pathogenesis and as treatment targets. Curr. Hypertens. Rep. 2010, 12, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.M.; Brosnan, M.J.; Graham, D.; Morton, J.J.; McPhaden, A.R.; McIntyre, M.; Hamilton, C.A.; Reid, J.L.; Dominiczak, A.F. Vascular smooth muscle cell polyploidy and cardiomyocyte hypertrophy due to chronic NOS inhibition in vivo. Am. J. Physiol. 1998, 274, H52–H59. [Google Scholar] [PubMed]
- Rudic, R.D.; Shesely, E.G.; Maeda, N.; Smithies, O.; Segal, S.S.; Sessa, W.C. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodeling. J. Clin. Investig. 1998, 101, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Structure of cerebral arterioles in mice deficient in expression of the gene for endothelial nitric oxide synthase. Circ. Res. 2004, 95, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Pappas, P.J.; Hobson, R.W., 2nd; Boric, M.P.; Sessa, W.C.; Durán, W.N. Endothelial nitric oxide synthase regulates microvascular hyperpermeability in vivo. J. Physiol. 2006, 574, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Faraci, F.M.; Sigmund, C.D.; Shesely, E.G.; Maeda, N.; Heistad, D.D. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am. J. Physiol. 1998, 274, H564–H570. [Google Scholar] [PubMed]
- Mashimo, H.; Goyal, R.K. Lessons from genetically engineered animal models. IV. Nitric oxide synthase gene knockout mice. Am. J. Physiol. 1999, 277, G745–G750. [Google Scholar] [PubMed]
- Fleming, I.; Busse, R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R1–R12. [Google Scholar] [CrossRef] [PubMed]
- Searles, C.D. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 2006, 291, C803–C816. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.P.; Kontos, H.A.; Christman, C.W.; DeWitt, D.S.; Povlishock, J.T. Superoxide generation and reversal of acetylcholine-induced cerebral arteriolar dilation after acute hypertension. Circ. Res. 1985, 57, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ceriello, A.; Giugliano, D.; Quatraro, A.; Lefebvre, P.J. Antioxidants shown an antihypertensive effect in diabetic and hypertensive subjects. Clin. Sci. 1991, 81, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Nakazono, K.; Watanabe, N.; Matsuno, J.; Saski, J.; Sato, T.; Inoue, M. Does superoxide underlie the pathogenesis of hypertension? Proc. Natl. Acad. Sci. USA 1991, 88, 10045–10048. [Google Scholar] [CrossRef] [PubMed]
- Pagano, P.J.; Chanock, S.J.; Siwik, D.A.; Colucci, W.S.; Clark, J.K. Angiotensin II induces p67phox mRNA expression and NADPH oxidase superoxide generation in rabbit aortic adventitial fibroblasts. Hypertension 1998, 32, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.E.; Rey, F.E.; Carretero, O.A.; Pagano, P.J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2234–H2240. [Google Scholar] [PubMed]
- Hsich, E.; Segal, B.H.; Pagano, P.J.; Rey, F.E.; Paigen, B.; Deleonardis, J.; Hoyt, R.F.; Holland, S.M.; Finkel, T. Vascular effects following homozygous disruption of p47(phox): An essential component of NADPH oxidase. Circulation 2000, 101, 1234–1236. [Google Scholar] [CrossRef] [PubMed]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(-) and systolic blood pressure in mice. Circ. Res. 2001, 89, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Faraci, F.M. Effects of NADH and NADPH on superoxide levels and cerebral vascular tone. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H688–H695. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, F.; Yang, X.P.; Jankowski, M.; Pagano, P.J. NAD(P)H oxidase mediates angiotensin II-induced vascular macrophage infiltration and medial hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Faraci, F.M. Angiotensin II produces superoxide-mediated impairment of endothelial function in cerebral arterioles. Stroke 2003, 34, 2038–2042. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, K.; Yamada, H.; Iwata, K.; Jin, D.; Katsuyama, M.; Matsuki, M.; Takai, S.; Yamanishi, K.; Miyazaki, M.; Matsubara, H.; et al. Nox1 is involved in angiotensin II-mediated hypertension: A study in Nox1-deficient mice. Circulation 2005, 112, 2677–2685. [Google Scholar] [CrossRef] [PubMed]
- Grote, K.; Ortmann, M.; Salguero, G.; Doerries, C.; Landmesser, U.; Luchtefeld, M.; Brandes, R.P.; Gwinner, W.; Tschernig, T.; Brabant, E.G.; et al. Critical role for p47phox in renin-angiotensin system activation and blood pressure regulation. Cardiovasc. Res. 2006, 71, 596–605. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, G.; Banfi, B.; Deffert, C.; Fiette, L.; Schappi, M.; Herrmann, F.; Krause, K.H. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. 2006, 580, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Nazarewicz, R.R.; Bikineyeva, A.; Hilenski, L.; Lassègue, B.; Griendling, K.K.; Harrison, D.G.; Dikalova, A.E. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid. Redox Signal. 2014, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Nickel, T.; Weis, M. Role of cytokines in cardiovascular diseases: A focus on endothelial responses to inflammation. Clin. Sci. 2005, 108, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Dworakowski, R.; Alom-Ruiz, S.P.; Shah, A.M. NADPH oxidase-derived reactive oxygen species in the regulation of endothelial phenotype. Pharmacol. Rep. 2008, 60, 21–28. [Google Scholar] [PubMed]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Sprague, A.H.; Khalil, R.A. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef] [PubMed]
- McMaster, W.G.; Kirabo, A.; Madhur, M.S.; Harrison, D.G. Inflammation, immunity, and hypertensive end-organ damage. Circ. Res. 2015, 116, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. eNOS uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res. 1994, 74, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Mohazzab, K.M.; Kaminski, P.M.; Wolin, M.S. NADH oxidoreductase is a major source of superoxide anion in bovine coronary artery endothelium. Am. J. Physiol. 1994, 266, H2568–H2572. [Google Scholar] [PubMed]
- Nguyen Dinh Cat, A.; Montezano, A.C.; Burger, D.; Touyz, R.M. Angiotensin II, NADPH oxidase, and redox signaling in the vasculature. Antioxid. Redox Signal. 2013, 19, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Vara, D.; Pula, G. Reactive oxygen species: Physiological roles in the regulation of vascular cells. Curr. Mol. Med. 2014, 14, 1103–1125. [Google Scholar] [CrossRef] [PubMed]
- Schnackenberg, C.G.; Welch, W.J.; Wilcox, C.S. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: Role of nitric oxide. Hypertension 1998, 32, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Di Wang, H.; Hope, S.; Du, Y.; Quinn, M.T.; Cayatte, A.; Pagano, P.J.; Cohen, R.A. Paracrine role of adventitial superoxide anion in mediating spontaneous tone of the isolated rat aorta in angiotensin II-induced hypertension. Hypertension 1999, 33, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Ryan, M.J.; Baumbach, G.L.; Sigmund, C.D.; Faraci, F.M. Superoxide contributes to vascular dysfunction in mice that express human renin and angiotensinogen. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1569–H1576. [Google Scholar] [CrossRef] [PubMed]
- Makino, A.; Skelton, M.M.; Zou, A.P.; Roman, R.J.; Cowley, A.W., Jr. Increased renalmedullary oxidative stress produces hypertension. Hypertension 2002, 39, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Lazartigues, E.; Lang, J.A.; Sinnayah, P.; Ahmad, I.M.; Spitz, D.R.; Davisson, R.L. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ. Res. 2002, 91, 1038–1045. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Lazartigues, E.; Sharma, R.V.; Davisson, R.L. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ. Res. 2004, 95, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Kinzenbaw, D.A.; Faraci, F.M. Critical role for CuZn-superoxide dismutase in preventing angiotensin II-induced endothelial dysfunction. Hypertension 2005, 46, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Ichiki, T.; Ito, K.; Takeshita, A. Induction of interleukin-6 expression by angiotensin II in rat vascular smooth muscle cells. Hypertension 1999, 34, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-kappaB transcription factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Kranzhöfer, R.; Schmidt, J.; Pfeiffer, C.A.; Hagl, S.; Libby, P.; Kübler, W. Angiotensin induces inflammatory activation of human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1623–1629. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Schieffer, E.; Hilfiker-Kleiner, D.; Hilfiker, A.; Kovanen, P.T.; Kaartinen, M.; Nussberger, J.; Harringer, W.; Drexler, H. Expression of angiotensin II and interleukin 6 in human coronary artery atherosclerotic plaques: Potential implications for inflammation and plaque instability. Circulation 2000, 101, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res. 2004, 94, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Schrader, L.I.; Kinzenbaw, D.A.; Johnson, A.W.; Faraci, F.M.; Didion, S.P. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2576–2581. [Google Scholar] [CrossRef] [PubMed]
- Gomolak, J.R.; Didion, S.P. Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front. Physiol. 2014, 5, 396. [Google Scholar] [CrossRef] [PubMed]
- Ala, Y.; Palluy, O.; Favero, J.; Bonne, C.; Modat, G.; Dornand, J. Hypoxia/reoxygenation stimulates endothelial cells to promote interleukin-1 and interleukin-6 production. Effects of free radical scavengers. Agents Actions 1992, 37, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Maruyama, M.; Fujita, T.; Arai, N.; Hayashi, R.; Araya, J.; Matsui, S.; Yamashita, N.; Sugiyama, E.; Kobayashi, M. Reactive oxygen intermediates stimulate interleukin-6 production in human bronchial epithelial cells. Am. J. Physiol. 1999, 276, L900–L908. [Google Scholar] [PubMed]
- Sano, M.; Fukuda, K.; Sato, T.; Kawaguchi, H.; Suematsu, M.; Matsuda, S.; Koyasu, S.; Matsui, H.; Yamauchi-Takihara, K.; Harada, M.; et al. ERK and p38 MAPK, but not NF-kappaB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ. Res. 2001, 89, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Hirohata, S.; Kashiwado, T.; Itoh, K.; Ishii, H. Cytokine regulation of hemostatic property and IL-6 production of human endothelial cells. Inflammation 1992, 16, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Hooper, W.C.; Phillips, D.J.; Renshaw, M.A.; Evatt, B.L.; Benson, J.M. The up-regulation of IL-6 and IL-8 in human endothelial cells by activated protein C. J. Immunol. 1998, 161, 2567–2573. [Google Scholar] [PubMed]
- Marin, V.; Montero-Julian, F.A.; Grès, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef] [PubMed]
- Watson, C.; Whittaker, S.; Smith, N.; Vora, A.J.; Dumonde, D.C.; Brown, K.A. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin. Exp. Immunol. 1996, 105, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentartutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. IL-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef]
- Fielding, C.A.; McLoughlin, R.M.; McLeod, L.; Colmont, C.S.; Najdovska, M.; Grail, D.; Ernst, M.; Jones, S.A.; Topley, N.; Jenkins, B.J. IL-6 regulates neutrophil trafficking during acute inflammation via STAT3. J. Immunol. 2008, 181, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Lee, C.; Sun, H.; LeJeune, W.; Recinos, A., 3rd; Ju, X.; Spratt, H.; Guo, D.C.; Milewicz, D.; Tilton, R.G.; et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Investig. 2009, 119, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Furuyama, F.; Yokota, Y.; Omori, Y.; Sato, T.; Tsunoda, F.; Iso, Y.; Koba, S.; Geshi, E.; Katagiri, T.; et al. IL-6 mobilizes bone marrow-derived cells to the vascular wall, resulting in neointima formation via inflammatory effects. J. Atheroscler. Thromb. 2014, 21, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.H.; Schlidt, S.A.; Chandel, N.S.; Hynes, K.L.; Schumacker, P.T.; Gewertz, B.L. Endothelial permeability and IL-6 production during hypoxia: Role of ROS in signal transduction. Am. J. Physiol. 1999, 277, L1057–L1065. [Google Scholar] [PubMed]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Coles, B.; Fielding, C.A.; Rose-John, S.; Scheller, J.; Jones, S.A.; O’Donnell, V.B. Classic interleukin-6 receptor signaling and interleukin-6 trans-signaling differentially control angiotensin II-dependent hypertension, cardiac signal transducer and activator of transcription-3 activation, and vascular hypertrophy in vivo. Am. J. Pathol. 2007, 171, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Ju, X.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A., 3rd; Brasier, A.R.; Tilton, R.G. Aortic adventitial fibroblasts participate in angiotensin-induced vascular wall inflammation and remodeling. J. Vasc. Res. 2011, 48, 261–272. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, S.; Ciechomska, M.; Cant, R.; van Laar, J.M. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-β (TGF-β) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J. Biol. Chem. 2014, 289, 9952–9960. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Jones, G.W.; McLoughlin, R.M.; McLeod, L.; Hammond, V.J.; Uceda, J.; Williams, A.S.; Lambie, M.; Foster, T.L.; Liao, C.T.; et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 2014, 40, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Castell, J.V.; Andus, T. Interleukin-6 and the acute phase response. Biochem. J. 1990, 265, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. The soluble interleukin-6 receptor and related proteins. Best Prac. Res. Clin. Endocrinol. Metab. 2015, 29, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Chava, K.R.; Karpurapu, M.; Wang, D.; Bhanoori, M.; Kundumani-Sridharan, V.; Zhang, Q.; Ichiki, T.; Glasgow, W.C.; Roa, G.N. CREB-mediated IL-6 expression is required for 15(S)-hydroxyeicosatetraenoic acid-induced vascular smooth muscle cell migration. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Sturgis, L.C.; Cannon, J.G.; Schreihofer, D.A.; Brands, M.W. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1742–R1748. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin II induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, K.; Ichiki, T.; Matsuura, H.; Miyazaki, R.; Hashimoto, T.; Takeda, K.; Sunagawa, K. Resveratrol attenuates angiotensin II-induced interleukin-6 expression and perivascular fibrosis. Hypertens. Res. 2009, 32, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Browatzki, M.; Schmidt, J.; Kübler, W.; Kranzhöfer, R. Endothelin-1 induces interleukin-6 release via activation of the transcription factor NF-kappaB in human vascular smooth muscle cells. Basic Res. Cardiol. 2000, 95, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Stankova, J.; Rola-Pleszczynski, M.; D’Orleans-Just, P. Endothelin 1 and thrombin synergistically stimulate IL-6 mRNA expression and protein production in human umbilical vein endothelial cells. J. Cardiovasc. Pharmacol. 1995, 26, S505–S507. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.P.; Liu, J.T.; Liu, N.; Guo, F.; Ji, Y.Y.; Pang, X. Pro-inflammatory effect of fibrinogen and FDP on vascular smooth muscle cells by IL-6, TNF-α and iNOS. Life Sci. 2011, 88, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Delneste, Y.; Lassalle, P.; Jeannin, P.; Joseph, M.; Tonnel, A.B.; Gosset, P. Histamine induces IL-6 production by human endothelial cells. Clin. Exp. Immunol. 1994, 98, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chi, L.; Stechschulte, D.J.; Dileepan, K.N. Histamine-induced production of interleukin-6 and interleukin-8 by human coronary artery endothelial cells is enhanced by endotoxin and tumor necrosis factor-alpha. Microvasc. Res. 2001, 61, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jin, M.; Hu, X.S.; Zhu, J.H. Homocysteine stimulates nuclear factor kappaB activity and interleukin-6 expression in rat vascular smooth muscle cells. Cell Biol. Int. 2006, 30, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Tritto, I.; Pinsky, D.; Liao, H.; Huang, J.; Fuller, G.; Brett, J.; May, L.; Stern, D. Induction of interleukin-6 (IL-6) by hypoxia in vascular cells. J. Biol. Chem. 1995, 270, 11463–11471. [Google Scholar] [CrossRef] [PubMed]
- May, L.T.; Torcia, G.; Cozzolino, F.; Ray, A.; Tatter, S.B.; Santhanam, U.; Sehgal, P.B.; Stern, D. Interleukin-6 gene expression in human endothelial cells: RNA start sites, multiple IL-6 proteins and inhibition of proliferation. Biochem. Biophys. Res. Commun. 1989, 159, 991–998. [Google Scholar] [CrossRef]
- Jirik, F.R.; Podor, T.J.; Hirano, T.; Kishimoto, T.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J. Immunol. 1989, 142, 144–147. [Google Scholar] [PubMed]
- Podor, T.J.; Jirik, F.R.; Loskutoff, D.J.; Carson, D.A.; Lotz, M. Human endothelial cells produce IL-6. Lack of response to exogenous IL-6. Ann. N. Y. Acad. Sci. 1989, 557, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Breviario, F.; Propserpio, P.; Biondi, A.; Vecchi, A.; van Damme, J.; Dejana, E.; Mantovani, A. IL-1 stimulates IL-6 production in endothelial cells. J. Immunol. 1989, 142, 549–553. [Google Scholar] [PubMed]
- Loppnow, H.; Libby, P. Adult human vascular endothelial cells express the IL6 gene differentially in response to LPS or IL1. Cell. Immunol. 1989, 122, 493–503. [Google Scholar] [CrossRef]
- Loppnow, H.; Libby, P. Comparative analysis of cytokine induction in human vascular endothelial and smooth muscle cells. Lymphokine Res. 1989, 8, 293–299. [Google Scholar] [PubMed]
- Loppnow, H.; Libby, P. Proliferating or interleukin 1-activated human vascular smooth muscle cells secrete copious interleukin 6. J. Clin. Investig. 1990, 85, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Norioka, K.; Hara, M.; Harigai, M.; Kitani, A.; Hirose, T.; Hirose, W.; Suzuki, K.; Kawakami, M.; Kawagoe, M.; Nakamura, H. Pretreatment of human vascular smooth muscle cells with interleukin-1 enhances interleukin-6 production and cell proliferation (action of IL-1 on vascular smooth muscle cells). Autoimmunity 1990, 7, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Howells, G.; Pham, P.; Taylor, D.; Foxwell, B.; Feldmann, M. Interleukin 4 induces interleukin 6 production by endothelial cells: Synergy with interferon-gamma. Eur. J. Immunol. 1991, 21, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Lee, W.H.; Kim, P.H. Oxidative mechanisms of IL-4-induced IL-6 expression in vascular endothelium. Cytokine 2010, 49, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Colotta, F.; Sironi, M.; Borrè, A.; Luini, W.; Maddalena, F.; Mantovani, A. Interleukin 4 amplifies monocyte chemotactic protein and interleukin 6 production by endothelial cells. Cytokine 1992, 4, 24–28. [Google Scholar] [CrossRef]
- Chen, C.C.; Manning, A.M. TGF-beta 1, IL-10 and IL-4 differentially modulate the cytokine-induced expression of IL-6 and IL-8 in human endothelial cells. Cytokine 1996, 8, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenberg, J.F.; von Asmuth, E.J.; Jeunhomme, T.M.; Buurman, W.A. IFN-gamma regulates the expression of the adhesion molecule ELAM-1 and IL-6 production by human endothelial cells in vitro. J. Immunol. 1990, 145, 2110–2114. [Google Scholar] [PubMed]
- Chi, L.; Li, Y.; Stehno-Bittel, L.; Gao, J.; Morrison, D.C.; Stechschulte, D.J.; Dileepan, K.N. Interleukin-6 production by endothelial cells via stimulation of protease-activated receptors is amplified by endotoxin and tumor necrosis factor-alpha. J. Interferon Cytokine Res. 2001, 21, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Jehle, A.B.; Li, Y.; Stechschulte, A.C.; Stechschulte, D.J.; Dileepan, K.N. Endotoxin and mast cell granule proteases synergistically activate human coronary artery endothelial cells to generate interleukin-6 and interleukin-8. J. Interferon Cytokine Res. 2000, 20, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Von Asmuth, E.J.; Leeuwenberg, J.F.; Ceska, M.; Buurman, W.A. LPS and cytokine-induced endothelial cell IL-6 release and ELAM-1 expression; involvement of serum. Eur. Cytokine Netw. 1991, 2, 291–297. [Google Scholar] [PubMed]
- Sasamoto, A.; Nagino, M.; Kobayashi, S.; Naruse, K.; Nimura, Y.; Sokabe, M. Mechanotransduction by integrin is essential for IL-6 secretion from endothelial cells in response to uniaxial continuous stretch. Am. J. Physiol. Cell Physiol. 2005, 288, C1012–C1022. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Nagino, M.; Komatsu, S.; Naruse, K.; Nimura, Y.; Nakanishi, M.; Sokabe, M. Stretch-induced IL-6 secretion from endothelial cells requires NF-kappaB activation. Biochem. Biophys. Res. Commun. 2003, 308, 306–312. [Google Scholar] [CrossRef]
- Kawai, M.; Naruse, K.; Komatsu, S.; Kobayashi, S.; Nagino, M.; Nimura, Y.; Sokabe, M. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: Clinical and experimental studies. J. Hepatol. 2002, 37, 240–246. [Google Scholar] [CrossRef]
- Zampetaki, A.; Zhang, Z.; Hu, Y.; Xu, Q. Biomechanical stress induces IL-6 expression in smooth muscle cells via Ras/Rac1-p38 MAPK-NF-kappaB signaling pathways. Am. J. Heart Circ. Physiol. 2005, 288, H2946–H2954. [Google Scholar] [CrossRef] [PubMed]
- Lubrano, V.; Baldi, S.; Ferrannini, E.; L’Abbate, A.; Natali, A. Role of thromboxane A2 receptor on the effects of oxidized LDL on microvascular endothelium nitric oxide, endothelin-1, and IL-6 production. Microcirculation 2008, 15, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, U.; Ikeda, M.; Oohara, T.; Oguchi, A.; Kamitani, K.; Tsuruya, Y.; Kano, S. Interleukin-6 stimulates the growth of vascular cells in a PDGF-dependent manner. Am. J. Physiol. 1991, 260, H1713–H1717. [Google Scholar] [PubMed]
- Gaumond, F.; Fortin, D.; Stankova, J.; Rola-Pleszczynski, M. Differential signaling pathways in platelet-activating factor-induced proliferation and interleukin-6 production by rat vascular smooth muscle cells. J. Cardiovasc. Pharmacol. 1997, 30, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ikeda, U.; Shimpo, M.; Yamamoto, K.; Shimada, K. Serotonin increases interleukin-6 synthesis in human vascular smooth muscle cells. Circulation 2000, 102, 2522–2527. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, S.; Inagaki, Y.; Nakamura, K.; Abe, R.; Shimizu, T.; Yoshimura, A.; Imaizumi, T. Pigment epithelium-derived factor inhibits TNF-alpha-induced interleukin-6 expression in endothelial cells by suppressing NADPH oxidase-mediated reactive oxygen species generation. J. Mol. Cell. Cardiol. 2004, 37, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Colston, J.T.; Chandrasekar, B.; Freeman, G.L. A novel peroxide-induced calcium transient regulates interleukin-6 expression in cardiac-derived fibroblasts. J. Biol. Chem. 2002, 277, 23477–23483. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochem. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Mihara, M.; Hashizume, M.; Yoshida, H.; Suzuki, M.; Shiina, M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clin. Sci. 2012, 122, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Yoshida, K.; Hibi, M.; Taga, T.; Kishimoto, T. Molecular cloning of a murine IL-6 receptor-associated signal transducer, gp130, and its regulated expression in vivo. J. Immunol. 1992, 148, 4066–4071. [Google Scholar] [PubMed]
- Demyanets, S.; Huber, K.; Wojta, J. Vascular effects of glycoprotein130 ligands- part II: Biomarkers and therapeutic targets. Vascul. Pharmacol. 2012, 57, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Klouche, M.; Bhakdi, S.; Hemme, M.; Rose-John, S. Novel path to activation of vascular smooth muscle cells: Up-regulation of gp130 creates an autocrine activation loop by IL-6 and its soluble receptor. J. Immunol. 1999, 163, 4583–4589. [Google Scholar] [PubMed]
- Boulanger, M.J.; Chow, D.C.; Brevnova, E.E.; Garcia, K.C. Hexameric structure and assembly of the interleukin-6/IL-6 α-receptor/gp130 complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Jenkins, B.J. Acquiring signalling specificity from the cytokine receptor gp130. Trends Genet. 2004, 20, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Schöbitz, B.; Pezeshki, G.; Pohl, T.; Hemmann, U.; Heinrich, P.C.; Holsboer, F.; Reul, J.M. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J. 1995, 9, 659–664. [Google Scholar] [PubMed]
- Garbers, C.; Aparicio-Siegmund, S.; Rose-John, S. The IL-6/gp130/STAT3 signaling axis: Recent advances towards specific inhibition. Curr. Opin. Immunol. 2015, 34, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Rousseau, F.; Guilhot, F.; Malinge, P.; Magistrelli, G.; Herren, S.; Jones, S.A.; Jones, G.W.; Scheller, J.; Lissilaa, R.; et al. Novel Insights into Interleukin 6 (IL-6) Cis- and trans-signaling pathways by differentially manipulating the assembly of the IL-6 signaling complex. J. Biol. Chem. 2015, 290, 26943–26953. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Jänner, N.; Chalaris, A.; Moss, M.L.; Floss, D.M.; Meyer, D.; Koch-Nolte, F.; Rose-John, S.; Scheller, J. Species specificity of ADAM10 and ADAM17 proteins in interleukin-6 (IL-6) trans-signaling and novel role of ADAM10 in inducible IL-6 receptor shedding. J. Biol. Chem. 2011, 286, 14804–14811. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, N.; Meyer, D.; Mauermann, A.; von der Heyde, J.; Wolf, J.; Schwarz, J.; Knittler, K.; Murphy, G.; Michalek, M.; Garbers, C.; et al. Shedding of Endogenous Interleukin-6 Receptor (IL-6R) Is Governed by A Disintegrin and Metalloproteinase (ADAM) Proteases while a Full-length IL-6R Isoform Localizes to Circulating Microvesicles. J. Biol. Chem. 2015, 290, 26059–26071. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.; Jacobs, S.; Ehlers, M.; Vollmer, P.; Mullberg, J.; Wolf, E.; Brem, G.; Meyer zum Buschenfelde, K.H.; Rose-John, S. The function of the soluble interleukin 6 (IL-6) receptor in vivo: Sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J. Exp. Med. 1996, 183, 1399–1406. [Google Scholar] [CrossRef] [PubMed]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Förster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling in vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Babon, J.J.; Varghese, L.N.; Nicola, N.A. Inhibition of IL-6 family cytokines by SOCS3. Semin. Immunol. 2014, 26, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, J.; Weissenbach, M.; Haan, S.; Heinrich, P.C.; Schaper, F. SOCS3 exerts its inhibitory function on interleukin-6 signal transduction through the SHP2 recruitment site of gp130. J. Biol. Chem. 2000, 275, 12848–12856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Badgwell, D.B.; Bevers, J.J., 3rd; Schlessinger, K.; Murray, P.J.; Levy, D.E.; Watowich, S.S. IL-6 signaling via the STAT3/SOCS3 pathway: Functional analysis of the conserved STAT3 N-domain. Mol. Cell. Biochem. 2006, 288, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [PubMed]
- Yasukawa, K.; Futatsugi, K.; Saito, T.; Yawata, H.; Narazaki, M.; Suzuki, H.; Taga, T.; Kishimoto, T. Association of recombinant soluble IL-6-signal transducer, gp130, with a complex of IL 6 and soluble IL-6 receptor, and establishment of an ELISA for soluble gp130. Immunol. Lett. 1992, 31, 123–130. [Google Scholar] [CrossRef]
- Wolf, J.; Waetzig, G.H.; Chalaris, A.; Reinheimer, T.M.; Wege, H.; Rose-John, S.; Garbers, C. Different Soluble Forms of the Interleukin-6 Family Signal Transducer gp130 Fine-tune the Blockade of Interleukin-6 Trans-signaling. J. Biol. Chem. 2016, 291, 16186–16196. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Thaiss, W.; Jones, G.W.; Waetzig, G.H.; Lorenzen, I.; Guilhot, F.; Lissilaa, R.; Ferlin, W.G.; Grötzinger, J.; Jones, S.A.; et al. Inhibition of classic signaling is a novel function of soluble glycoprotein 130 (sgp130), which is controlled by the ratio of interleukin 6 and soluble interleukin 6 receptor. J. Biol. Chem. 2011, 286, 42959–42970. [Google Scholar] [CrossRef] [PubMed]
- Müller-Newen, G.; Küster, A.; Hemmann, U.; Keul, R.; Horsten, U.; Martens, A.; Graeve, L.; Wijdenes, J.; Heinrich, P.C. Soluble IL-6 receptor potentiates the antagonistic activity of soluble gp130 on IL-6 responses. J. Immunol. 1998, 161, 6347–6355. [Google Scholar] [PubMed]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Kovaleva, M.; Rabe, B.; Eichler, J.; Kallen, K.J.; Rose-John, S. Development of a monoclonal antibody-based enzyme-linked immunoabsorbent assay for the binding of gp130 to the IL-6/IL-6R complex and its competitive inhibition. J. Immunol. Methods 2004, 291, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Naka, T.; Nishimoto, N.; Kishimoto, T. The paradigm of IL-6: From basic science to medicine. Arthritis Res. 2002, 4, S233–S242. [Google Scholar] [CrossRef] [PubMed]
- Eckes, B.; Hunzelmann, N.; Ziegler-Heitbrock, L.Z.; Urbanski, A.; Luger, T.; Krieg, T.; Mauch, C. Interleukin-6 expression by fibroblasts grown in three-dimensional gel culture. FEBS Lett. 1992, 298, 229–232. [Google Scholar] [CrossRef]
- Song, Y.; Shen, H.; Schenten, D.; Shan, P.; Lee, P.J.; Goldstein, D.R. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Wung, B.S.; Hsu, M.C.; Wu, C.C.; Hsieh, C.W. Resveratrol suppresses IL-6-induced ICAM-1 gene expression in endothelial cells: Effects on the inhibition of STAT3 phosphorylation. Life Sci. 2005, 78, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Wung, B.S.; Ni, C.W.; Wang, D.L. ICAM-1 induction by TNFalpha and IL-6 is mediated by distinct pathways via Rac in endothelial cells. J. Biomed. Sci. 2005, 12, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Kaplanski, G.; Marin, V.; Montero-Julian, F.; Mantovani, A.; Farnarier, C. IL-6: A regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003, 24, 25–29. [Google Scholar] [CrossRef]
- Saura, M.; Zaragoza, C.; Bao, C.; Herranz, B.; Rodriguez-Puyol, M.; Lowenstein, C.J. Stat3 mediates interleukin-6 inhibition of human endothelial nitric-oxide synthase expression. J. Biol. Chem. 2006, 281, 30057–30062. [Google Scholar] [CrossRef] [PubMed]
- Hung, M.J.; Cherng, W.J.; Hung, M.Y.; Wu, H.T.; Pang, J.H. Interleukin-6 inhibits endothelial nitric oxide synthase activation and increases endothelial nitric oxide synthase binding to stabilized caveolin-1 in human vascular endothelial cells. J. Hypertens. 2010, 28, 940–951. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.I.; Chen, X.; Didion, S.P. Heterozygous eNOS deficiency is associated with oxidative stress and endothelial dysfunction in diet-induced obesity. Physiol. Rep. 2015, 3, e12630. [Google Scholar] [CrossRef] [PubMed]
- Luckett, L.R.; Gallucci, R.M. Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br. J. Dermatol. 2007, 156, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, B.; Luchtefeld, M.; Braun, S.; Hilfiker, A.; Hilfiker-Kleiner, D.; Drexler, H. Role of NAD(P)H oxidase in angiotensin II-induced JAK/STAT signaling and cytokine induction. Circ. Res. 2000, 87, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Fujibayashi, M.; Fujiwara, Y.; Kaneko, T.; Xia, C.; Imai, E.; Kamada, T.; Ando, A.; Ueda, N. Angiotensin II stimulates interleukin-6 release from cultured mouse mesangial cells. J. Am. Soc. Nephrol. 1995, 6, 95–101. [Google Scholar] [PubMed]
- Satou, R.; Gonzalez-Villalobos, R.A.; Miyata, K.; Ohashi, N.; Katsurada, A.; Navar, L.G.; Kobori, H. Costimulation with angiotensin II and interleukin 6 augments angiotensinogen expression in cultured human renal proximal tubular cells. Am. J. Physiol. Ren. Physiol. 2008, 295, F283–F289. [Google Scholar] [CrossRef] [PubMed]
- Satou, R.; Gonzalez-Villalobos, R.A.; Miyata, K.; Ohashi, N.; Urushihara, M.; Acres, O.W.; Navar, L.G.; Kobori, H. IL-6 augments angiotensinogen in primary cultured renal proximal tubular cells. Mol. Cell. Endocrinol. 2009, 311, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Nabata, T.; Morimono, S.; Koh, E.; Shiraishi, T.; Ogihara, T. Interleukin-6 stimulates c-myc expression and proliferation of cultured vascular smooth muscle cells. Biochem. Int. 1990, 20, 445–453. [Google Scholar] [PubMed]
- Wang, Z.; Newman, W.H. Smooth muscle cell migration stimulated by interleukin 6 is associated with cytoskeletal reorganization. J. Surg. Res. 2003, 111, 261–266. [Google Scholar] [CrossRef]
- Johnson, A.W.; Kinzenbaw, D.A.; Modrick, M.L.; Faraci, F.M. Small-molecule inhibitors of signal transducer and activator of transcription 3 protect against angiotensin II-induced vascular dysfunction and hypertension. Hypertension 2013, 61, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Birukov, K.G. Cyclic stretch, reactive oxygen species, and vascular remodeling. Antioxid. Redox Signal. 2009, 11, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Lemarié, C.A.; Tharaux, P.L.; Lehoux, S. Extracellular matrix alterations in hypertensive vascular remodeling. J. Mol. Cell. Cardiol. 2010, 48, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Briones, A.M.; Arribas, S.M.; Salaices, M. Role of extracellular matrix in vascular remodeling of hypertension. Curr. Opin. Nephrol. Hypertens. 2010, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.A.; Chatterjee, A.; Mitra, A.; Pathak, K.; Mahata, S.K.; Sarkar, S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 2012, 287, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, Y.; Jia, L.; Han, Y.; Cheng, J.; Li, H.; Qi, Y.; Du, J. Macrophage-stimulated cardiac fibroblast production of IL-6 is essential for TGF β/Smad activation and cardiac fibrosis induced by angiotensin II. PLoS ONE 2012, 7, e35144. [Google Scholar] [CrossRef] [PubMed]
- Recinos, A., 3rd; LeJeune, W.S.; Sun, H.; Lee, C.Y.; Tieu, B.C.; Lu, M.; Hou, T.; Boldogh, I.; Tilton, R.G.; Brasier, A.R. Angiotensin II induces IL-6 expression and the Jak-STAT3 pathway in aortic adventitia of LDL receptor-deficient mice. Atherosclerosis 2007, 194, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-β signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, R.M.; Lee, E.G.; Tomasek, J.J. IL-6 modulates alpha-smooth muscle actin in dermal fibroblasts from IL-6-deficient mice. J. Investig. Dermatol. 2006, 126, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.Q.; Kondo, T.; Ishida, Y.; Takayasu, T.; Mukaida, N. Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 2003, 73, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Bhagat, K.; Vallance, P. Inflammatory cytokines impair endothelium-dependent dilatation in human veins in vivo. Circulation 1997, 96, 3042–3047. [Google Scholar] [CrossRef] [PubMed]
- Orshal, J.M.; Khalil, R.A. Interleukin-6 impairs endothelium-dependent NO-cGMP-mediated relaxation and enhances contraction in systemic vessels of pregnant rats. Am. J. Regul. Integr. Comp. Physiol. 2004, 286, R1013–R1023. [Google Scholar] [CrossRef] [PubMed]
- Orshal, J.M.; Khalil, R.A. Reduced endothelial NO-cGMP-mediated vascular relaxation and hypertension in IL-6-infused pregnant rats. Hypertension 2004, 43, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Chae, C.U.; Lee, R.T.; Rifai, N.; Ridker, P.M. Blood pressure and inflammation in apparently healthy men. Hypertension 2001, 38, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Gibas-Dorna, M.; Nowak, D.; Piatek, J.; Pupek-Musialik, D.; Krauss, H.; Kopczynski, P. Plasma ghrelin and interleukin-6 levels correlate with body mass index and arterial blood pressure in males with essential hypertension. J. Physiol. Pharmacol. 2015, 66, 367–372. [Google Scholar] [PubMed]
- Naya, M.; Tsukamoto, T.; Morita, K.; Katoh, C.; Furumoto, T.; Fujii, S.; Tamaki, N.; Tsutsui, H. Plasma interleukin-6 and tumor necrosis factor-alpha can predict coronary endothelial dysfunction in hypertensive patients. Hypertens. Res. 2007, 30, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Vayreda, M.; Richart, C.; Gutierrez, C.; Broch, M.; Vendrell, J.; Ricart, W. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. J. Clin. Endocrinol. Metab. 2001, 86, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Preiser, J.C.; Schmartz, D.; Van der Linden, P.; Content, J.; Vanden Bussche, P.; Buurman, W.; Sebald, W.; Dupont, E.; Pinsky, M.R.; Vincent, J.L. Interleukin-6 administration has no acute hemodynamic or hematologic effect in the dog. Cytokine 1991, 3, 1–4. [Google Scholar] [CrossRef]
- Boesen, E.I.; Pollock, D.M. Effect of chronic IL-6 infusion on acute pressor responses to vasoconstrictors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1745–H1749. [Google Scholar] [CrossRef] [PubMed]
- Melendez, G.C.; McLarty, J.L.; Levick, S.P.; Du, Y.; Janicki, J.S.; Brower, G.L. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 2010, 56, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Suematsu, S.; Matsuda, T.; Aozasa, K.; Akira, S.; Nakano, N.; Ohno, S.; Miyazaki, J.I.; Yamamura, K.I.; Hirano, T.; Kishimoto, T. IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc. Natl. Acad. Sci. USA 1989, 86, 7547–7551. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.K.; Syrkina, O.L.; Kolliputi, N.; Mark, E.J.; Hales, C.A.; Waxman, A.B. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 2009, 104, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Fattori, E.; Della Rocca, C.; Costa, P.; Giorgio, M.; Dente, B.; Pozzi, D.L.; Ciliberto, G. Development of progressive kidney damage and myeloma kidney in interleukin-6 transgenic mice. Blood 1994, 83, 2570–2579. [Google Scholar] [PubMed]
- Kovalchuk, A.L.; Kim, J.S.; Park, S.S.; Coleman, A.E.; Ward, J.M.; Morse, H.C., 3rd; Kishimoto, T.; Potter, M.; Janz, S. IL-6 transgenic mouse model for extraosseous plasmacytoma. Proc. Natl. Acad. Sci. USA 2002, 99, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A.; Nakagawa, P.; Liu, Y.; Leung, P.; Dai, X.; Yang, X.P.; Peterson, E.L.; Carretero, O.A. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J. Hypertens. 2015, 33, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Leite, R.; Fleming, C.; Pollock, J.S.; Webb, R.C.; Brands, M.W. Hypertensive response to acute stress is attenuated in interleukin-6 knockout mice. Hypertension 2004, 44, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.L.; Stergis, L.C.; Labazi, H.; Osborne, J.B., Jr.; Fleming, C.; Pollock, J.S.; Manhiani, M.; Imig, J.D.; Brands, M.W. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am. J. Heart Circ. Physiol. 2006, 290, H935–H940. [Google Scholar] [CrossRef] [PubMed]
- Brands, M.W.; Banes-Berceli, A.K.; Inscho, E.W.; Al-Azawi, H.; Allen, A.J.; Labazi, H. Interleukin 6 knockout prevents angiotensin II hypertension: Role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 2010, 56, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, W.; Yu, H.; Zhang, Y.; Dai, Y.; Ning, C.; Tao, L.; Sun, H.; Kellems, R.E.; Blackburn, M.R.; et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension 2012, 59, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Manhiani, M.M.; Seth, D.M.; Banes-Berceli, A.K.; Satou, R.; Navar, L.G.; Brands, M.W. The role of IL-6 in the physiological versus hypertensive blood pressure actions of angiotensin II. Physiol. Rep. 2015, 3, e12595. [Google Scholar] [CrossRef] [PubMed]
- Manhiani, M.M.; Quigley, J.E.; Socha, M.J.; Motamed, K.; Imig, J.D. IL6 supression provides renal protection independent of blood pressure in a murine model of salt-sensitive hypertension. Kidney Blood Press. Res. 2007, 30, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ijaz, T.; Sun, H.; Ray, S.; Lejeune, W.; Lee, C.; Recinos, A., 3rd; Guo, D.C.; Milewicz, D.M.; Tilton, R.G.; et al. Interleukin-6-signal transducer and activator of transcription-3 signaling mediates aortic dissections induced by angiotensin II via the T-helper lymphocyte 17-interleukin 17 axis in C57BL/6 mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Rateri, D.L.; Cassis, L.A.; Daugherty, A. Subcutaneous angiotensin II infusion using osmotic pumps induces aortic aneurysms in mice. J. Vis. Exp. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zouein, F.A.; Zgheib, C.; Hamza, S.; Fuseler, J.W.; Hall, J.E.; Soljancic, A.; Lopez-Ruiz, A.; Kurdi, M.; Booz, G.W. Role of STAT3 in angiotensin II-induced hypertension and cardiac remodeling revealed by mice lacking STAT3 serine 727 phosphorylation. Hypertens. Res. 2013, 36, 496–503. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Stimulus | Reference |
|---|---|
| 15(S)-HETE | [80] |
| Activated Protein C | [63] |
| Aldosterone | [81] |
| Angiotensin II | [52,53,58,74,83] |
| Endothelin | [84,85] |
| Fibrinogen | [86] |
| Histamine | [87,88] |
| Homocysteine | [89] |
| Hydrogen Peroxide | [61,116] |
| Hypoxia | [59,71,90] |
| Interleukin-1α/β | [94,95,96,97,98] |
| Interleukin-4 | [99,100,101,102] |
| Interferon-γ | [98,103] |
| Lipopolysaccharide | [92,93,94,95,104,105,106] |
| Mechanical Stretch | [107,108,109,110] |
| Oxidized LDL | [111] |
| Platelet-derived Growth Factor | [112,113] |
| Serotonin | [114] |
| Thrombin | [86] |
| Tumor Necrosis Factor-α | [92,96,104,115] |
| Basal Blood Pressure | Reference | |
|---|---|---|
| IL-6 Infusion | No effect | [177,178,179] |
| IL-6-Deficiency | No effect | [57,73,81,184,185,186,187,188,189] |
| Model | Hypertension Development | Reference |
|---|---|---|
| Acute Stress | Partial Inhibition | [185] |
| Ang II | No Effect | [57,185] |
| Partial Inhibition | [73,187,188] | |
| Ang II + High-salt | No Effect | [184,190] |
| Partial Inhibition | [186] | |
| Ang II + RKM | No Effect | [189] |
| DOCA-salt | No Effect | [81] |
| High-salt | No Effect | [184,186] |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. https://doi.org/10.3390/ijms18122563
Didion SP. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. International Journal of Molecular Sciences. 2017; 18(12):2563. https://doi.org/10.3390/ijms18122563
Chicago/Turabian StyleDidion, Sean P. 2017. "Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature" International Journal of Molecular Sciences 18, no. 12: 2563. https://doi.org/10.3390/ijms18122563
APA StyleDidion, S. P. (2017). Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. International Journal of Molecular Sciences, 18(12), 2563. https://doi.org/10.3390/ijms18122563